Proactively designing viral diagnostics at scale with machine learning
Published in Bioengineering & Biotechnology

When you take a test for COVID-19, you expect the diagnosis to be right. You base the protection of your health, as well as the health of the friends and family you come into contact with, on that COVID test. It’s vital that the diagnostic is both sensitive (if you have COVID, it will tell you that you do) so you can stop spread and seek treatment if needed, and that it’s specific (if you don’t have COVID, it will tell you that you don’t) so you aren’t unnecessarily isolating or so you can seek an alternative diagnosis.
While we’re used to relying on COVID tests, many don’t realize that diagnostic design is “somewhat of an art, and not fully predictable.” For example, PCR test design must satisfy a large variety of criteria, often requiring manual assessment based on in-depth knowledge of its thermodynamics. However, in a quick response to a novel disease outbreak, it’s easy for some of these checks to slip through the cracks, causing design flaws. This happened with the US CDC’s initial COVID diagnostic panel, discussed in detail here. Even when these checks are satisfied, we do not have clear standards to compare different designs’ sensitivity or specificity as neither are easily predictable. For viruses with greater variation than SARS-CoV-2, such as influenza, finding a diagnostic that works across all common strains is even more difficult.
Our lab, the Sabeti Lab at the Broad Institute MIT and Harvard, is dedicated to improving the state of infectious disease monitoring, in part by developing novel diagnostic platforms with several close partners based on CRISPR-Cas13a technology, which is meant to reduce the time and cost of testing. Like standard PCR diagnostics, CRISPR-Cas13a detects viral nucleic acids. First, we amplify a small section of the viral genome. Then, a small CRISPR RNA guide binds to a part of that section, activating Cas13a enzymes that go on to produce a measurable signal. SHINE, the point of care platform for CRISPR-Cas13a, provides PCR quality performance with the ease-of-use of a rapid test. CARMEN, a multiplexed test using CRISPR-Cas13a, enables testing hundreds of viruses simultaneously, which can, for example, help to distinguish between respiratory viruses with similar symptoms but different treatments.

ADAPT’s web interface for SARS-CoV-2 assay designs.
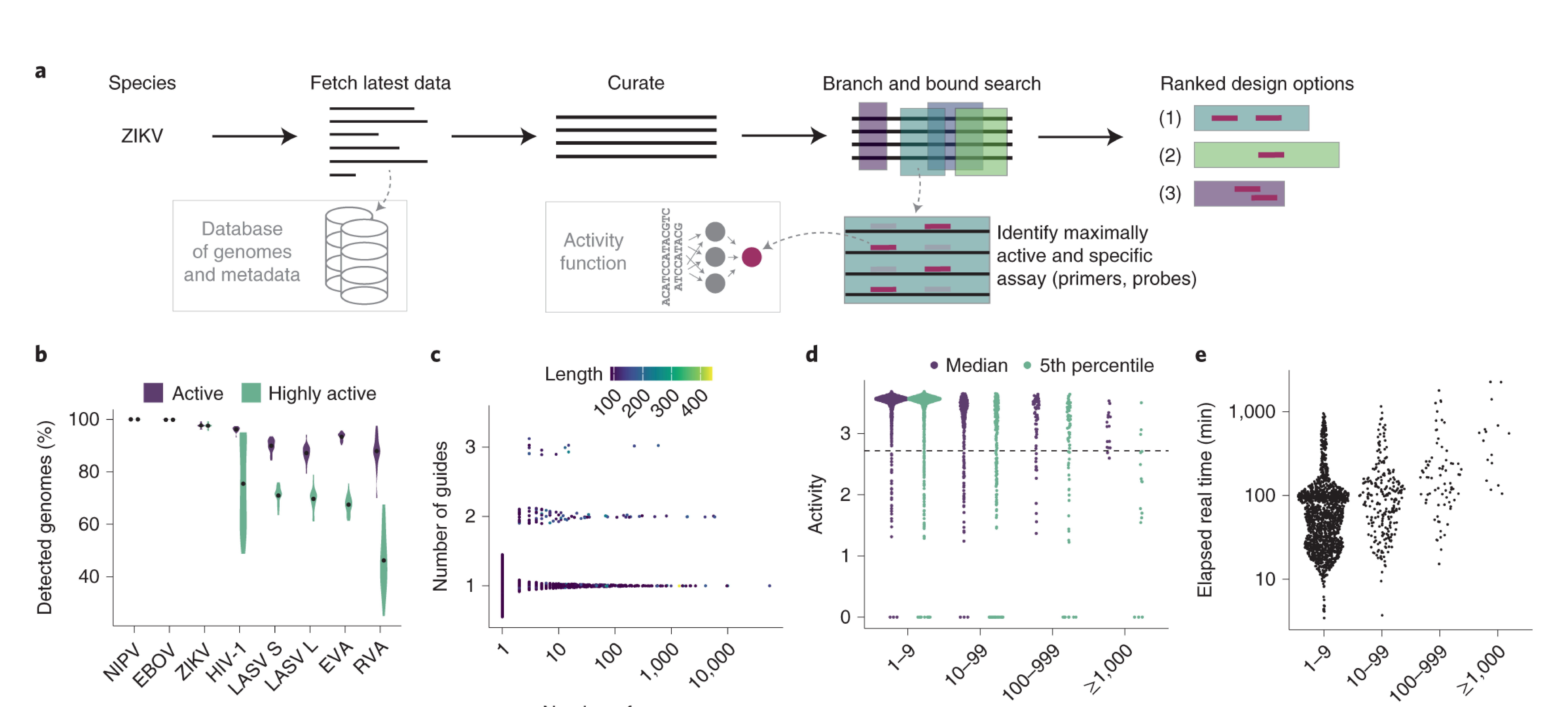
We need a tool to design assays for these technologies that accounts for their unique biochemistry, detects the range of diversity within a virus, and does not mistakenly detect other viruses. It also ought to be able to generalize to other diagnostic technologies like PCR with the same goals. We thus created a method and built ADAPT to quickly and automatically design diagnostics to be both sensitive and specific.
To be sensitive, methods of targeting pathogen genomes—whether it be for diagnostic, therapy, or vaccine countermeasures—must account for both potency and breadth. Potency is how strong the activity is; breadth is how robust it is to variation in the genome, such as across different variants. It is common for methods to prioritize, or even focus entirely on, only one of these two goals. Potency is typically measured against a single reference viral strain and ignoring all other strains of the virus. Breadth is often prioritized by only targeting highly conserved regions of the genome. A major goal of our work is to consider these simultaneously and find a balance between these two. Our previous work on CATCH, a method for designing a set of probes that would capture a wide range of microbial genomic diversity for sequencing purposes, addressed a similar problem, and so we were curious if a related approach could work here for viral diagnostics.

To account for the potency dimension of sensitivity for CRISPR-Cas13a diagnostics—that is, ensure we can detect low levels of the virus—we focused on creating a model for the enzymatic activity of the RNA guide, the part that can be programmed to detect particular viral sequences. Prior to our method, there was no quantitative model to predict the enzymatic activity of CRISPR RNA guides for Cas13a when detecting a viral target based on their sequence and the mismatches they may have with the target; the standard for design has been heuristic rules. Such a quantitative model could help us to rank and prioritize different guide sequences. We developed a two-part hurdle model, which represents the biochemistry as first requiring a guide to bind (the “hurdle”) and then producing a measurable signal. The “hurdle” is a classification model that produces a binary output of whether or not the guide produces any signal or not. A regression model follows and determines the rate at which the guide will produce fluorescence, which is a proxy for how low of a viral load it’s able to detect. We tested a number of different underlying models and found convolutional neural networks with an additional locally connected layer performed best for both the classification and regression model. This is potentially because the convolutional filters capture position-independent effects, such as multiple mismatches right next to each other that can cause the guide to fail no matter where they occur. The locally connected layer additionally captures position-dependent effects, such as a particular region that requires a certain sequence composition.
To account for the breadth dimension of sensitivity, we efficiently consider all known viral variation and, using our predictive model, design assays to perform well across that variation. First, we automatically download and align viral genomes from NCBI’s viral genome database. Then, we use this dataset to generate potential guides at each location in the alignment and assess them. The overall objective is to maximize enzymatic activity, or the potency of our diagnostic, across the variation in the dataset. We find regions that can be amplified, and then, at each position in these regions, we find the combination of guides that has the maximum average predicted activity, penalizing assays with too many guides or that might be too hard to amplify.
While we aimed for our assays to have both potency and breadth to sensitively capture all virus strains we intend to target, we still wanted to make sure our assays were specific—we did not want them to detect other viruses. We use the same automatic system to download the genomes of closely related viruses, break their sequences into easily searchable chunks, and check assays against them in a way that accounts for RNA binding dynamics.
Through this method, ADAPT is a computationally powerful tool, but even the best tools can fail if they are hard to use or aren't updated. To alleviate this possibility, we created a user-friendly website for ADAPT.
In accordance with our larger goal to prepare for and quickly respond to outbreaks, we provide a resource of pre-designed sensitive and specific assays for over a thousand vertebrate-infecting viruses (mostly at the species level), to capture the viruses that not only already infect humans but also have a high likelihood to jump from infecting another vertebrate to infecting humans. Since viruses are constantly evolving and emerging, we created a pipeline to regularly update this resource by running ADAPT in parallel on a cloud computing cluster, allowing it to keep pace with the newest variation.

We know that this resource doesn’t account for every scenario ADAPT is able to handle, so we included a user interface for running ADAPT on custom data or custom settings using our computational resources on the Amazon Web Services cloud. Through this website, ADAPT is accessible even for people who are unfamiliar with the command line or who don’t have access to sufficient computational resources. For both pre-designed and custom assay designs, the ADAPT website provides an intuitive visualization of the assay with biologically relevant context, including a summary of its predicted performance, its location in the genome relative to important genomic features, and a measurement of genomic variance at each position.
ADAPT’s core methods are broadly applicable to the challenge of targeting diverse genomes while avoiding unintended targets. We are currently working on applying ADAPT to other diagnostic technologies, like PCR, as design for these diagnostic technologies could also benefit from these methods. Sequence-based antivirals and vaccines could also use these approaches. We are also interested in expanding ADAPT to track and assess older, widely-used assays so that we can warn if these assays may fail on the newest emerging variants. At the same time, we are working to forecast what variants may emerge so that we can account for them during the design process.
ADAPT can help us not only proactively design assays for the next outbreak, but also for new variants emerging during the current pandemic. In addition to being an accurate and accessible method for designing CRISPR-based diagnostics, ADAPT is a framework for diagnostic design more generally and can be a powerful tool for multiple technologies. We hope ADAPT can support the scientific and public health communities in robustly detecting emerging and evolving viruses, thereby helping to prevent the next outbreak.
Thanks to Jon Aritzi Sanz, Lily Chylek, & Pardis C. Sabeti for reviewing this post.
Follow the Topic
-
Nature Biotechnology
A monthly journal covering the science and business of biotechnology, with new concepts in technology/methodology of relevance to the biological, biomedical, agricultural and environmental sciences.


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in