Putting the pieces together: Developing a new tool to comprehensively update the Mycobacterium tuberculosis H37Rv reference sequence
Published in Microbiology

Mycobacterium tuberculosis (M. tuberculosis), the causative agent of the disease Tuberculosis (TB), is estimated to infect roughly a quarter of the world’s population. The World Health Organization (WHO) estimated that in 2020, TB was the second leading cause of death from an infectious disease after SARS-CoV-2. The ability to track and study M. tuberculosis outbreaks relies heavily on accurate Whole Genome Sequencing (WGS). In 1998, Stewart Cole and colleagues published the first complete genome of M. tuberculosis, strain H37Rv, using a painstaking Bacterial Artificial Chromosome (BAC) and Cosmid sequencing approach. The work was conducted by two independent trans-Atlantic groups working on different isolates of H37Rv. This work ushered in a new era of TB research utilizing the lab adapted H37Rv strain with its genome widely accepted as the M. tuberculosis reference sequence.
In the 20 odd years since the first M. tuberculosis genome was published, Next Generation Sequencing (NGS) and third generation sequencing approaches have significantly improved our ability to study M. tuberculosis genomes. This has been particularly useful given that M. tuberculosis is extremely GC rich and roughly 10% of its genome is comprised of highly repetitive PE/PPE genes. To overcome these inherent difficulties in studying M. tuberculosis genomes, we developed Bact-Builder, a novel pipeline for the de novo assembly of M. tuberculosis genomes. Bact-Builder was designed using a consensus genome + polishing approach (Fig 1a). Generating a consensus genome allows us to evaluate multiple assemblers and ensure that our final genome does not contain erroneous redundant sequences or missing regions. Polishing the consensus genome with both long and short read sequencing further allows us to correct for single nucleotide polymorphisms (SNPs) and indels that are notoriously found in homopolymeric regions.
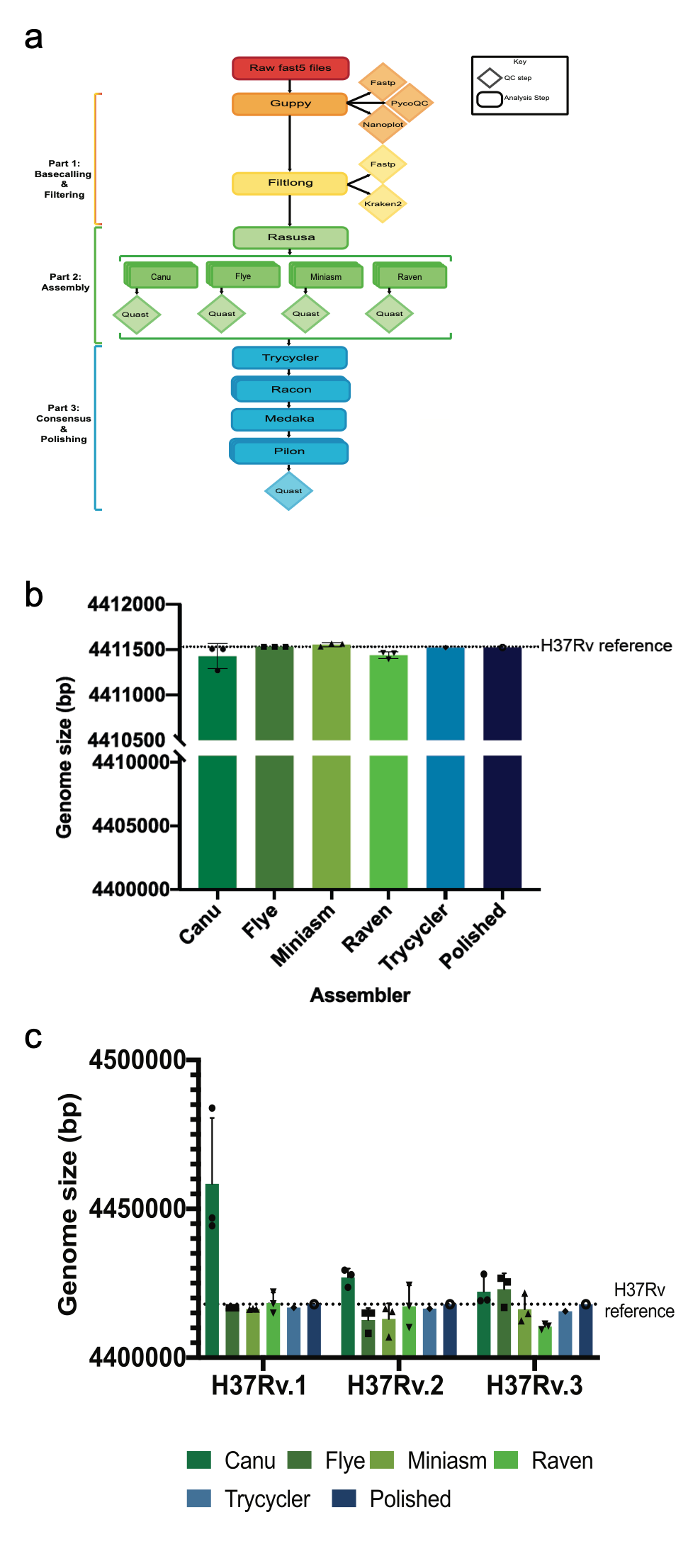
b. Comparing in silico H37Rv assemblies and
Bact-Builder output.
c. Comparing 3 replicates of in vitro
H37Rv and the Bact-Builder output
Bact-Builder was originally developed after evaluating assemblies with artificial in silico reads generated using the published 1998 H37Rv sequence (Fig 1a). Multiple assemblers run in triplicate using an artificial ideal data set demonstrated variability not only between assemblers but also within assemblers (Fig 1b). We observed that when we generated a consensus genome with Trycycler, variability disappeared, and further polishing produced a virtually identical genome to that of the published sequence (Fig 1b). We repeated these tests on actual samples of H37Rv and found the same phenomena after applying Bact-Builder to generate a polished consensus genome (Fig 1c).
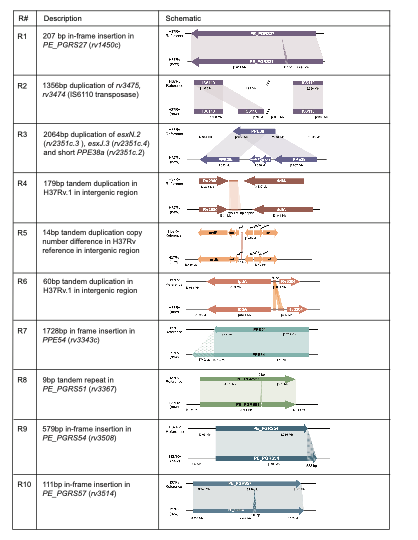
We took this one step further and compared the output of our in vitro Bact-Builder data to the published reference and identified 109 SNPs, 35 indels, and 10 large regions of difference between the 2 sequences (Fig 2). Most of the regions of difference between our sequence and the 1998 sequence were in-frame insertions in the highly repetitive
PE/PPE genes, and differences in tandem duplications in intergenic regions. However, in region 3 (R3) we identified 2 new paralogs of esxN and esxJ (esxN.2 and esxJ.3) and a novel truncated duplication of PPE38 (PPE38a). These new paralogs are particularly interesting because they belong to the conserved ESX-5 loci which has been linked to PE/PPE protein secretion during macrophage infection. We further confirmed that not only were the novel paralogs actively transcribed, but their expression levels were different from the known copies of the gene suggesting a novel function for these paralogs. Further work to confirm the role of these paralogs is needed.
The real benefits of Bact-Builder are the potential downstream applications. Highly accurate de novo genomes will allow us to establish new references for in vivo studies, better understand lineage and strain differences, and enable accurate functional analysis. Translationally, this tool will allow us study M. tuberculosis outbreaks in the context they are occurring to better understand and treat one of the most dangerous pathogens in the world.
and H37Rv (new)Caption
Follow the Topic
-
Nature Communications
An open access, multidisciplinary journal dedicated to publishing high-quality research in all areas of the biological, health, physical, chemical and Earth sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Women's Health
Publishing Model: Hybrid
Deadline: Ongoing
Tumor Microenvironment Crosstalk and Therapeutic Implications
Publishing Model: Hybrid
Deadline: Nov 02, 2026


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in