SCCAF: machine learning infers putative cell types
Published in Protocols & Methods
Explore the Research
Putative cell type discovery from single-cell gene expression data - Nature Methods
SCCAF automates the discovery of putative cell types and their feature genes using scRNA-seq data.
Identifying cell types in multicellular organisms and understanding the relationships between them has been a major aim of biological research since the discovery of cells by Robert Hooke almost 400 years ago. Historically, cell types have been defined by their morphology as assessed by microscopy, by their locations in an organism, by their function in vivo or in vitro, by their and evolutionary history, or by the expression of a small number of molecular markers on the cell surface according to flow cytometry.
Data from single-cell RNA-sequencing is one of the most data-rich and high-dimensional sources of information for the discovery of new putative cell types and refining the classification of existing ones. In spite of many available computational analysis tools, the annotation of cell types (or cell states) according to - data on the expert inspections of cell clusters and marker gene expression. The data annotation is laborious and time-consuming, and is becoming a major bottleneck in high throughput projects, such as the Human Cell Atlas.
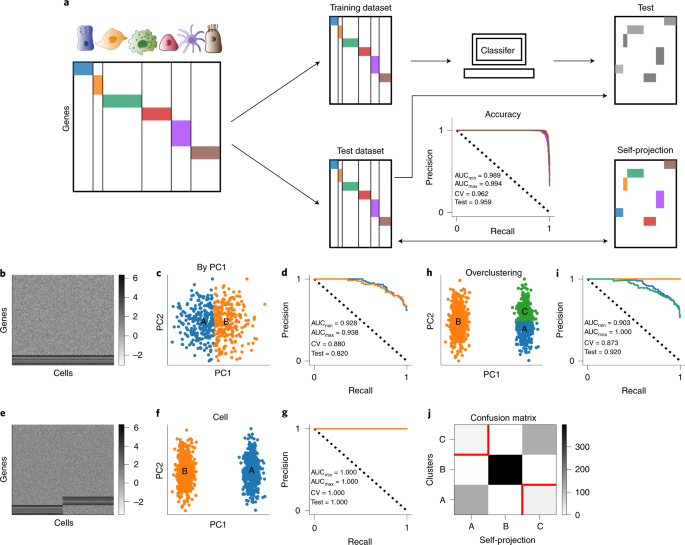
How about asking the computer to help us in defining the cell types and annotating the data? If a machine learning model can learn the biological signature of a cell type, perhaps it should be able to do a better job in assigning cell types for a large dataset than a human expert? If so, the machine learning model should encode the essential features of the cell types in the . When cells of the same type are clustered into multiple clusters, machine learning may be able to find that there is a “confusion” between cell types and the clusters. Such cell clusters can be considered as the “same cell type”.

But how to achieve a cell type-related clustering with little human intervention? We can start from an over-clustering status and use the machine learning confusion to merge the cell clusters and achieve the cell clusters. Here we propose an automated method that allows for a potential discovery of novel, not yet annotated putative cell types. Our method, which we call Single Cell Clustering Assessment Framework (SCCAF), is based on the iterative application of machine learning and self-projection to clusters (train and test a model on data split from the same dataset). This involves starting with small “overclustered” groups of cells, and then gradually merging the clusters that correspond to the same putative cell type. Finally, SCCAF a set of defining feature genes to each group of cells. Benchmarking on many well-annotated published datasets, SCCAF is able to restore results identical to manual annotations in most cases.
We aim to automatically identify biologically meaningful groups of cells that can be discriminated from all other cells via models based on the expression of appropriate feature genes. Our approach can be combined with reference-based methods, such as , Moana and , first to identify meaningful clusters, and then to annotate them, e.g., to assign names derived from earlier datasets. Our method is not only implemented as an open-source also has been developed as a part of our implementation of a high-throughput data analysis pipeline in the Galaxy tool kit and will be used in the EBI’s Single Cell Expression Atlas (www.ebi.ac.uk/gxz/sc).
I do RNA computational biology, trying to understand both the structural form of RNA molecules and RNA determined gene regulatory networks in cells.
Follow the Topic
-
Nature Methods
This journal is a forum for the publication of novel methods and significant improvements to tried-and-tested basic research techniques in the life sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Methods development in Cryo-ET and in situ structural determination
Publishing Model: Hybrid
Deadline: Oct 30, 2026
Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in