Seeing the Tree in a forest of microbial diversity
Published in Microbiology
“We know so much more than this!” – our initial reaction to search results for “Tree of Life”. Wikipedia has an entry, of course. We were happy to see the Tree of Life it presented was based on organisms with fully sequenced genomes. We were startled to see it was from 2006, when there was a grand total of 191 genomes available. This was the current public perception of life’s diversity?
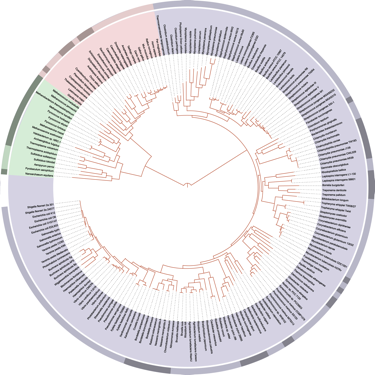
Wikipedia’s current view of the Tree of Life (from the Tree of Life (biology) entry;), with bacteria in blue, archaea in red and eukaryotes in green. Image credit: Ivica Letunic and Mariana Ruiz Villarreal, image in the public domain.
Wikipedia isn’t to blame. Between isolate sequencing, metagenomics, and single cell sequencing, we are now able to reconstruct genomes for organisms at an unprecedented rate. The numbers of new candidate phyla, and the abundance of their members across different environments have dramatically re-shaped our understanding of life’s variety. Tens of thousands of genomes have been sequenced, covering organisms from all three domains of life. The last few years have been exciting times in environmental microbiology, but a comprehensive view of this newly described diversity was missing.
It was time for an update, a Tree of Life for the genomic era.
We wanted our Tree of Life to cover the total diversity of life on Earth, from a genomic perspective. We used publicly available genomes from the Joint Genome Institute’s IMG database, as well as genomes from over a thousand organisms identified at a wide variety of sampling sites, totaling 3,083 genomes.

Environments surveyed whose organisms contributed over 1,000 previously unpublished genomes to our Tree of Life. Clockwise from top left: White Oak River estuary (credit: Dirk Frankenburg); Yellowstone National Park hot spring (credit: Dan Coleman); Rifle, Colorado aquifer (credit: Ken Williams); dolphin oral microbiome (credit: US Navy Marine Mammal Program); Soledad Formation in the Atacama Desert (credit: Kari Finstad); Crystal Geyser, Utah (credit: Chris Brown); Angelo Coast Range Reserve meadow, California (credit: Susan Spaulding); Honorobe Underground Research Center (credit: Japan Atomic Energy Agency).
We designed our dataset carefully to reduce some of the sampling biases present in genome collections. We selected one representative organism from each genus to include on the tree. For some relatively understudied phyla, this included almost every genome available (e.g., Deferribacteres, a phylum with seven sequenced genomes from seven different genera). For other lineages, this represented a massive reduction in genome numbers (i.e., there are ~4,500 Staphylococcus genomes available; we chose one).
We inferred our trees using the CIPRES portal, a powerful public resource for phylogenetic research. We built trees based on SSU RNA genes as well as based on a concatenated alignment of sixteen ribosomal protein genes. Both trees highlighted the wealth of biological diversity covered by genome sequences – not only do we know these organisms exist, but we also have access to the blueprints of their metabolic potential and ecosystem roles. This depth of understanding is particularly important in light of the amount of diversity that exists within currently uncultured lineages of microorganisms – organisms for which a genome sequence is the most direct information available.
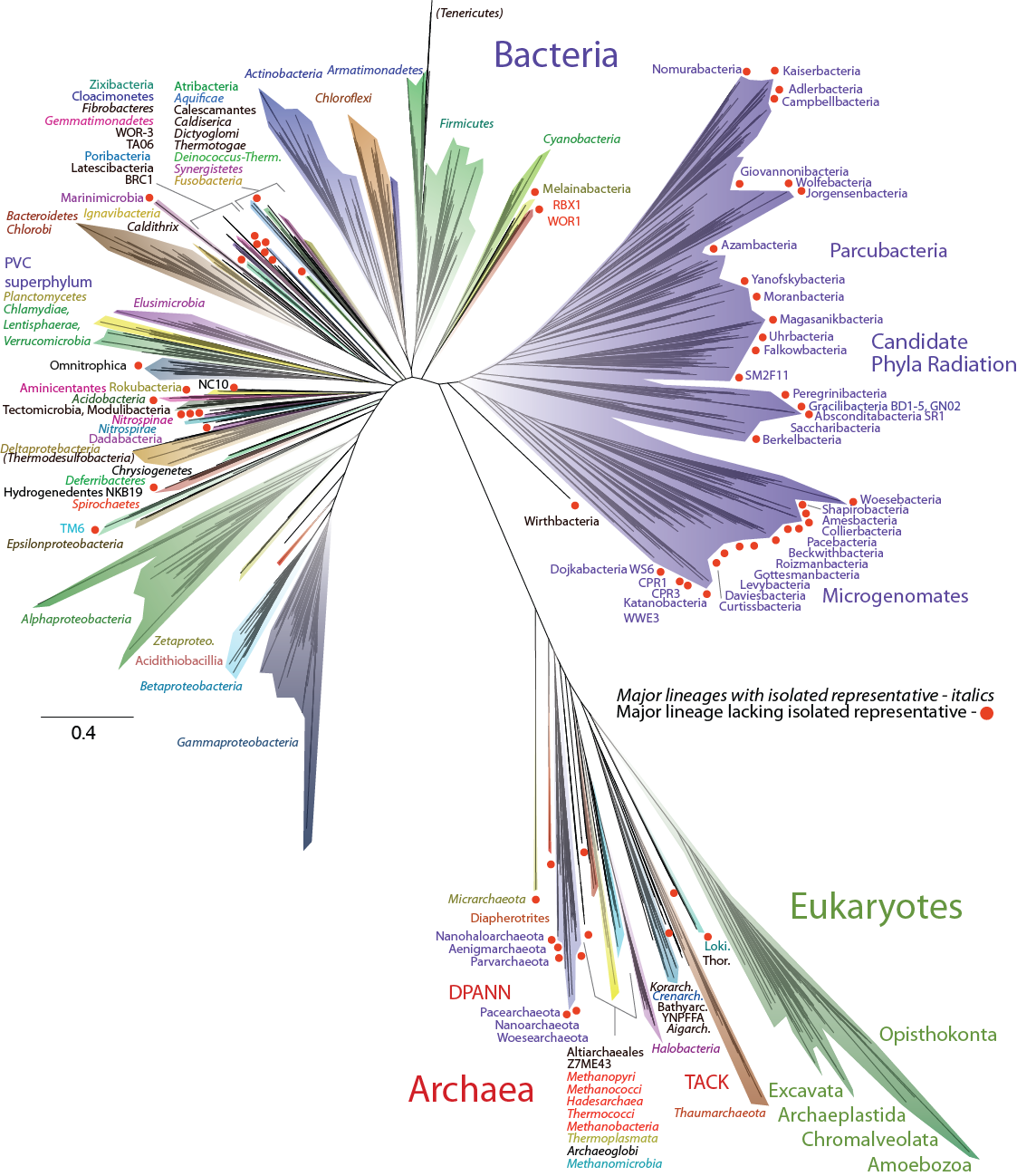
Comprehensive Tree of Life based on a concatenated alignment of sixteen ribosomal proteins. Major lineages are highlighted by colored wedges. Lineages lacking a cultivated representative are marked with red dots.
The SSU rRNA tree and the concatenated ribosomal tree showed different deep topologies: one with three domains (Bacteria, Archaea, and Eukaryotes), and one with two (where Eukaryotes branched out of Archaea). We weren’t trying to answer this deep evolutionary question with our datasets, and the differences in the two trees, and lack of support for those deeper relationships, emphasize that more information will be needed for this long-running debate to be resolved.
We were struck by how much of the tree was composed of lineages with no cultured representatives (red dots), and also how different the evolutionary distances within phyla seemed to be. Some phyla were quite small from a diversity perspective while others contained a huge amount. To see how well current taxonomy aligns with the underlying sequence divergence, we collapsed the tree at a uniform average branch length, so clades would all have the same approximate diversity within them. It was a telling exercise. Some well known phyla collapsed together into one clade (e.g., the Firmicutes, Actinobacteria, and Chloroflexi together), while others split into multiple clades (e.g., the Spirochaetes into four groups, and the Candidate Phylum Woesearchaeota into six). The Candidate Phyla Radiation (CPR) made up approximately 50% of the total bacterial diversity on the tree with this view.
Our tree presents a new view of the diversity of life, from a genome perspective. Exploration of new environments, and deeper sequencing of well-studied systems continue to uncover new organisms and lineages on the Tree, meaning the next decade in environmental microbiology promises to be exciting (and diverse) as well!


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in