Shortcut to Chemically Accurate Quantum Computing via Density-based Basis-set Correction

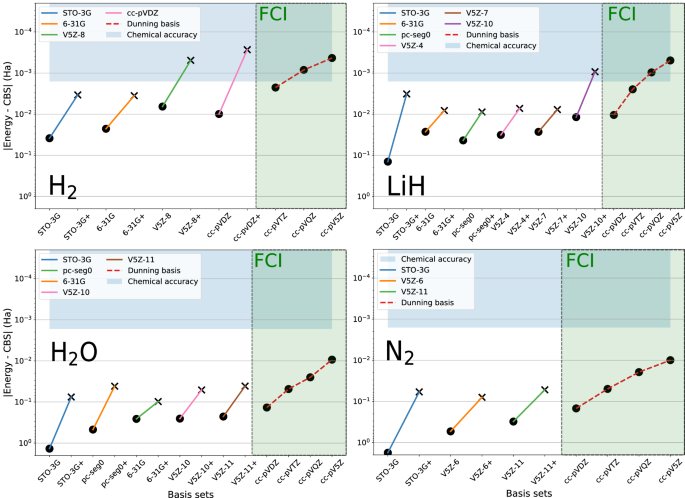
Quantum chemistry on classical computers suffers from the resource requirement which grows exponentially with the size of the chemical systems. Therefore, most of the methods focus on providing chemically accurate energy differences and molecular properties for systems with up to twenty light atoms and sometimes transition metals requiring expensive calculations on highly performant computers when available.
With the promise of quantum computing to be more suitable to represent chemical systems [Feynman, 1982] and to have better resource management than classical computation, quantum chemists can hope to see their field extended to more complex systems with industrial applications.
One natural question to ask here is why would someone access highly accurate electronic structure descriptions of industrially relevant systems when other efficient approximations have been developed for decades? Concerning quantum computing, the electronic Hamiltonian is expressed in second quantization, employing an encoding that maps one spin-orbital to one qubit. This mapping allows one to represent an exponentially large Hilbert space using only a linear number of qubits. However, to achieve accurate and practically valuable predictions for chemical systems in real-world applications, the molecular Hamiltonian should be expressed and solved using extensive basis sets of one-electron orbital functions. The number of qubits required for such calculations quickly exceeds the available capacities on current Noisy Intermediate Scale Quantum (NISQ) devices, upcoming early Fault-Tolerant Quantum Computing (FTQC) devices, and high-performance classical emulators.
With this context in mind, one can understand our recent publication Shortcut to Chemically Accurate Quantum Computing via Density-based Basis-set correction as being a set of novel strategies to manage task distribution between classical and quantum resources. We detail the following complementary possible paths of improvement.
First, we propose to exploit a strategy based on classical Density Functional Theory embedding of any quantum computing ansatz for chemistry. It is based on the density-based basis-set correction (DBSSC) [Giner, et al, 2018]. We introduce two DBBSC workflows. The first one is a self-consistent basis-set correction which iteratively updates the short-range electronic density using a functional of the density: the development of this strategy has been originally motivated by the fact that density-functional approximation tends to have a better convergence of the density with the basis-set size. In other terms, using DBBSC should reduce the qubit requirements of a given ansatz. The second workflow permits the use of a posteriori correction that can be added to any type of computations on real hardware, as long as the electronic density is available at the Hartree-Fock level which is the level of theory used to initialize the molecular orbitals and the operator pools. Therefore, the second workflow allows a more complete exploitation of the initial information (the Hartree-Fock density) available to us and necessary to initialize the quantum computation workflow.
Finally, all DBBSC schemes are coupled to a modified pivoted-Cholesky [Lehtola, 2019] strategy for an on-the-fly generation of basis sets based on the further exploitation of the Hartree-Fock computations. Such basis-sets are “system-adapted” (denoted SABS) since they are specifically adapted to a given molecular system providing a reduced size compared to the originate target basis set. They can find use in both classical- and quantum-computation for quantum chemists.
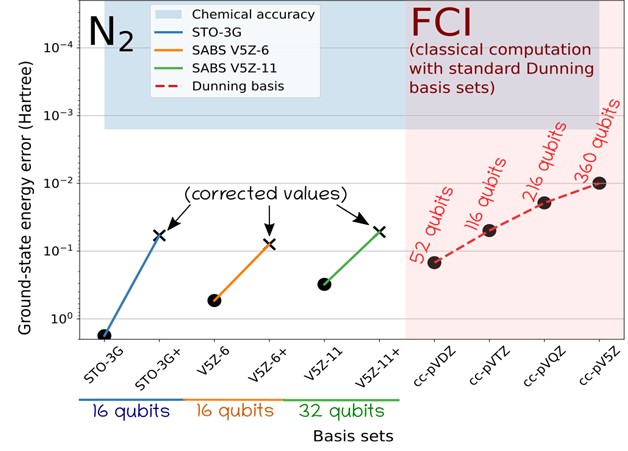
We apply the presented global strategies to ground-state energies, dipole moments and dissociation curves: we show how this strategy allows us to strongly reduce the qubit requirements. Using GPU-accelerated state-vector emulation, we are able to obtain quantitative quantum-chemistry results on molecules that would otherwise require brute-force quantum calculations using hundreds of logical qubits. It is important to note that such computations require a fairly reasonable amount of HPC resources. Our methodology allows us to compute chemically meaningful energies and properties on systems that would have required far more than 100 logical qubits. For example, the computation of the H2 total energy at the FCI/cc-pV5Z level that would have required more than 220 logical qubits, can be achieved here with only 24 qubits using our basis-set correction scheme and our SABS technique. Overall, we were able to converge four systems to the FCI/CBS limit including He, Be, H2, and LiH. We were also able to provide accurate dissociation curves for H2, LiH, and N2 (up to a triple zeta quality).
This research opens the path to more affordable quantitative quantum-chemistry simulations of small molecules using QC algorithms. We hope this work will be a cornerstone for new types of strategies for better resource management and, in a long-term perspective, to ease applicability of quantum computing for quantum chemistry of industrially relevant applications. To conclude, by reducing the number of qubits required to reach the CBS limit, we expect to tackle predictive real-world quantum chemistry applications with strategies applicable to both NISQ and FTQC algorithms.
References
[Quantum Energy Initiative] Quantum Energy Initiative, https://quantum-energy-initiative.org/. Accessed 30 August 2024.
[Cao, et al, 2018] Cao, Yudong, et al. “Potential of quantum computing for drug discovery.” IBM Journal of Research and Development, vol. 62, no. 6, 2018, pp. 6-1.
[Feynman, 1982] Feynman, Richard. “Simulating physics with computers.” International Journal of Theoretical Journals, vol. 21, no. 6, 1982, pp. 467-488.
[Giner, et al, 2018] Giner, Emmanuel, et al. “Curing basis-set convergence of wave-function theory using density-functional theory: a systematically improvable approach.” The Journal of Chemical Physics, vol. 149, 2018, p. 194301.
[Lehtola, 2019] Lehtola, Susi. “Curing basis set overcompleteness with pivoted Cholesky decompositions.” The Journal of Chemical Physics, vol. 151, no. 24, 2019.
Follow the Topic
-
Communications Chemistry
An open access journal from Nature Portfolio publishing high-quality research, reviews and commentary in all areas of the chemical sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Chemical modification of proteins
Publishing Model: Open Access
Deadline: Sep 30, 2026
Sustainable waste management through polymer upcycling
Publishing Model: Open Access
Deadline: Aug 31, 2026




Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in