Single-cell biological network inference using a heterogeneous graph transformer
Published in Protocols & Methods

Artificial intelligence (AI) and single-cell studies have been making waves in the science and technology communities. By sequencing the genetic material of one cell at a time, researchers can uncover the unique features and characteristics that make each cell in our body special. This single-cell sequencing technique has already revolutionized our understanding of the brain, cancer, and the immune system. AI offers a broad range of methods that can be used to investigate diverse data- and hypothesis-driven questions in single-cell biology. Specifically, the highly heterogeneous nature of single-cell sequencing data can be analyzed across a wide range of research topics by generalizing deep-learning model design and optimization. However, there is a catch. Each type of single-cell sequencing data only gives us a glimpse of what is happening inside a cell. It is like looking at a tiny piece of a puzzle - we can see some details but do not get the whole picture. That is where a new approach called single-cell multi-omics comes in. By analyzing multiple types of data from the same cell, researchers can better understand how cells work and what makes them unique.
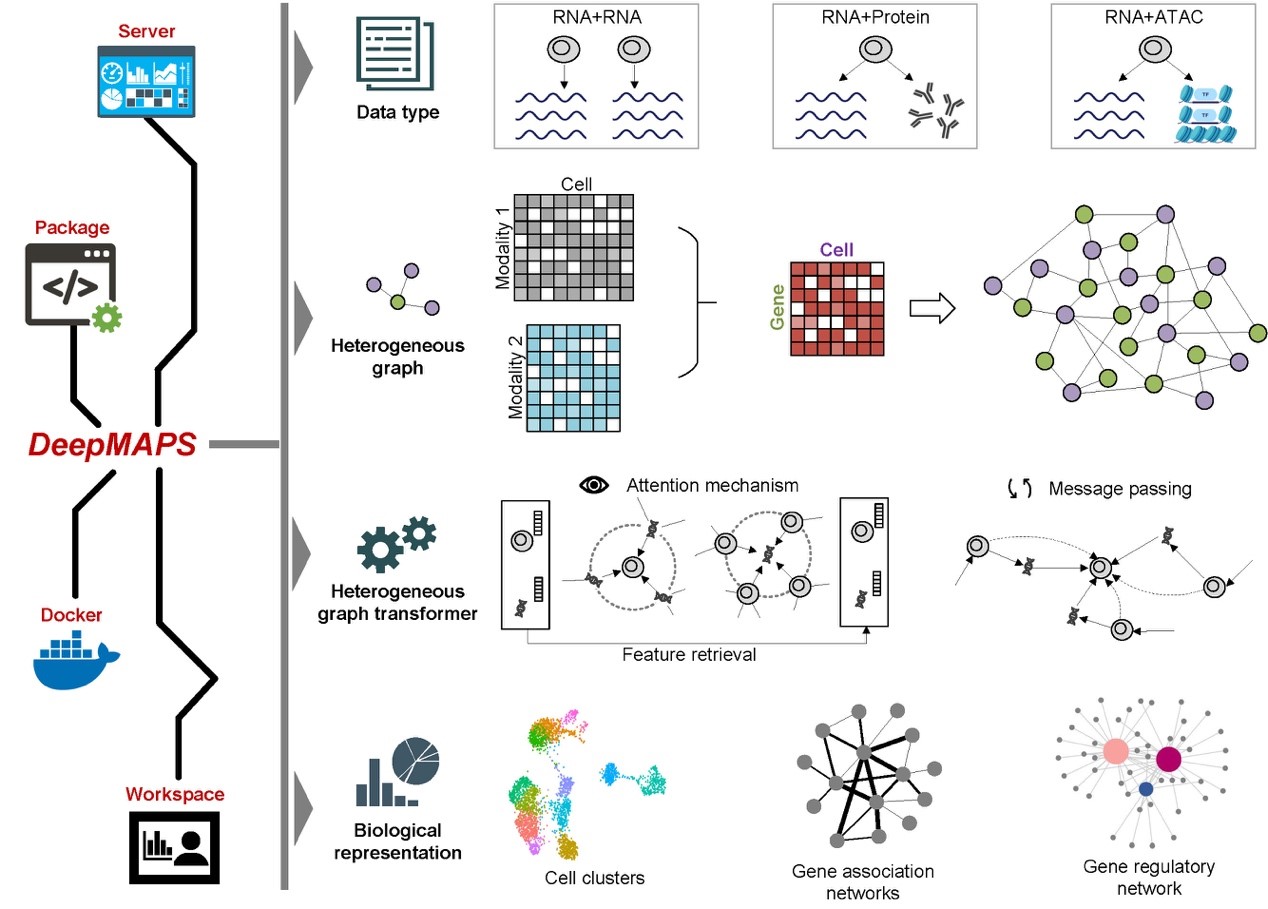
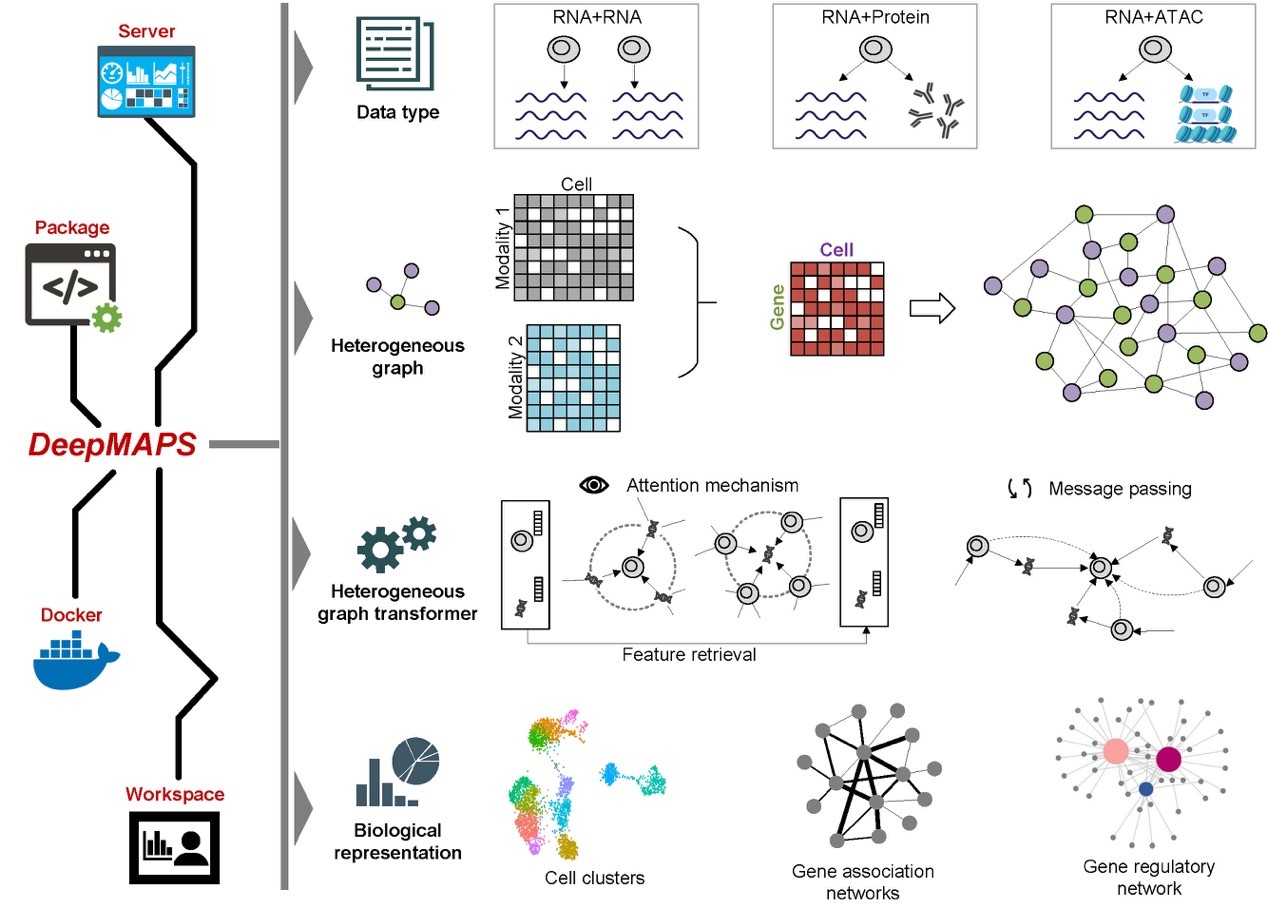
Several tools have been developed to help scientists make sense of the vast amounts of data generated by single-cell sequencing, e.g., Seurat, MOFA, and Harmony. These tools can predict what types of cells are present in a sample, remove unwanted variations between different batches of cells, and identify connections between different types of data. However, most of these tools do not consider the complex relationships between cells and the different types of data (e.g., genes, proteins, and enhancers) being analyzed. Graph neural network (GNN) is a type of deep learning algorithm good for modeling and analyzing single-cell sequencing data with a graph representation learning style. For example, this technique allows them to build a graph that connects different cells and genes based on their similarity. It can also identify the active biological networks that control how cells function.
DeepMAPS (Deep learning-based Multi-omics Analysis Platform for Single-cell data) is newly developed to help scientists analyze and interpret single-cell multi-omics data. It uses a cutting-edge GNN model, specifically the heterogeneous graph transformer (HGT), to model single-cell multi-omics data and identify the active biological networks that control cells' functions. Specifically, it creates a graph that includes both cells and genes as nodes, with their relationships acting as edges. The HGT model looks at all the connections between cells and genes to understand how they interact, without making any assumptions about which genes work together. One cool thing about HGT is that it can estimate which genes are most important for specific cells, which helps scientists understand how cells work and the underlying molecular program. Specifically, DeepMAPS outperforms other methods in terms of accuracy and efficiency, and it is effective at analyzing data from lung tumors and other types of cancers. To make DeepMAPS more accessible, we have also created a web server that provides a range of useful features and visualizations. With this tool, scientists can easily explore their data and gain new insights into the complex world of cellular biology. In addition, DeepMAPS can infer two biological networks: gene association and regulatory networks (GRN). To measure the importance of genes for cell function, we use "centrality scores" and "functional enrichment." In one study, we compared the gene association networks created by DeepMAPS to those created by other tools and found that the networks created by DeepMAPS were more closely connected and more relevant to cell function. Our research indicates how DeepMAPS can help us learn more about the genes and networks that make up our cells, and how they work together to keep us healthy.
Our team at the Bioinformatics and Mathematical Biosciences Lab (BMBL) of the Ohio State University focus on the research of single-cell multi-omics data and are very excited to deliver and further maintain DeepMAPS. Users can find the DeepMAPS source code at: https://github.com/OSU-BMBL/deepmaps. The web server is available at: https://bmblx.bmi.osumc.edu/. To read more about our approach and methods, check the DeepMAPS paper published on Nature Communications: https://www.nature.com/articles/s41467-023-36559-0. As a long-term goal, we aim to develop cutting-edge computational tools to discover underlying molecular mechanisms in diverse biological systems and complex diseases. For more information, visit https://u.osu.edu/bmbl/.
Follow the Topic
-
Nature Communications
An open access, multidisciplinary journal dedicated to publishing high-quality research in all areas of the biological, health, physical, chemical and Earth sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Women's Health
Publishing Model: Hybrid
Deadline: Ongoing
Tumor Microenvironment Crosstalk and Therapeutic Implications
Publishing Model: Hybrid
Deadline: Nov 02, 2026




Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in