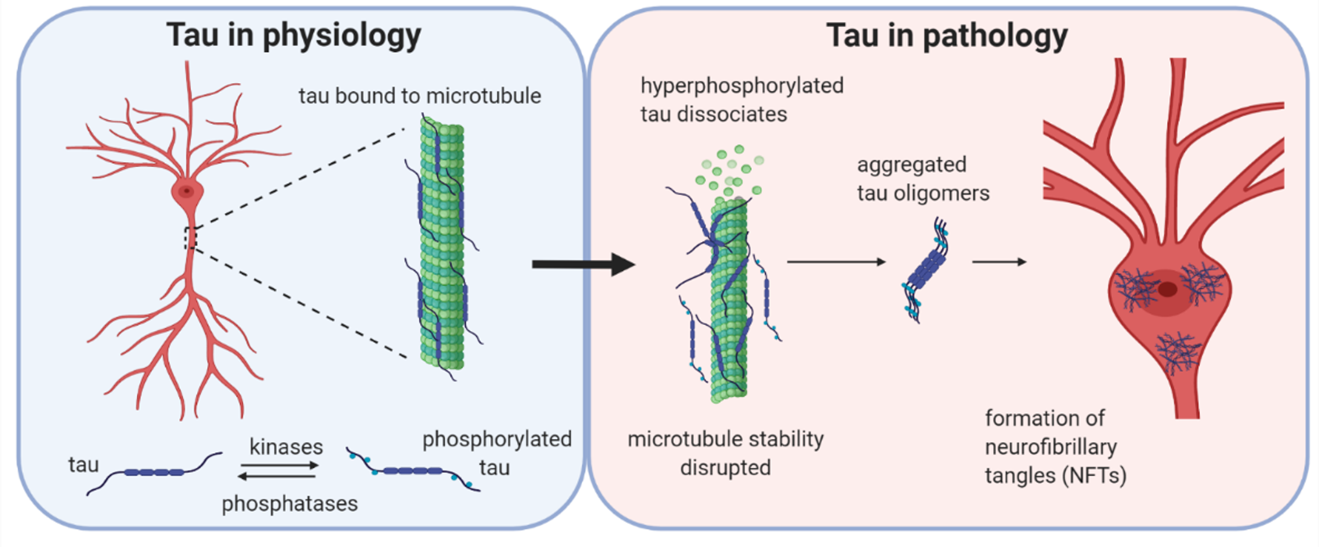

Tau is a microtubule-associated protein with many physiological functions, including modifying microtubule stability, neuronal morphology, vesicle transport and trafficking (1,2). Tau dysfunction contributes to a number of diseases, termed tauopathies, either as the primary causative agent (e.g., Pick’s disease) or as a component of the neuropathology (e.g., Alzheimer’s disease). In Alzheimer’s disease, abnormally phosphorylated tau dissociates from microtubules and aggregates to form oligomers and fibrils, accumulating in the soma-dendritic compartment. Tau can further aggregate to form neurofibrillary tangles (NFTs), whose abundance correlates with disease progression (3,4). However, it appears that the soluble tau oligomers (oTau) are the most bioactive, in terms of disrupting neuronal function. Indeed, the toxic effects of tau oligomers can occur in the absence of NFT pathology (5–7). Although the toxicity of oTau is now well established, the mechanistic basis of oTau actions on neuronal function are poorly understood.
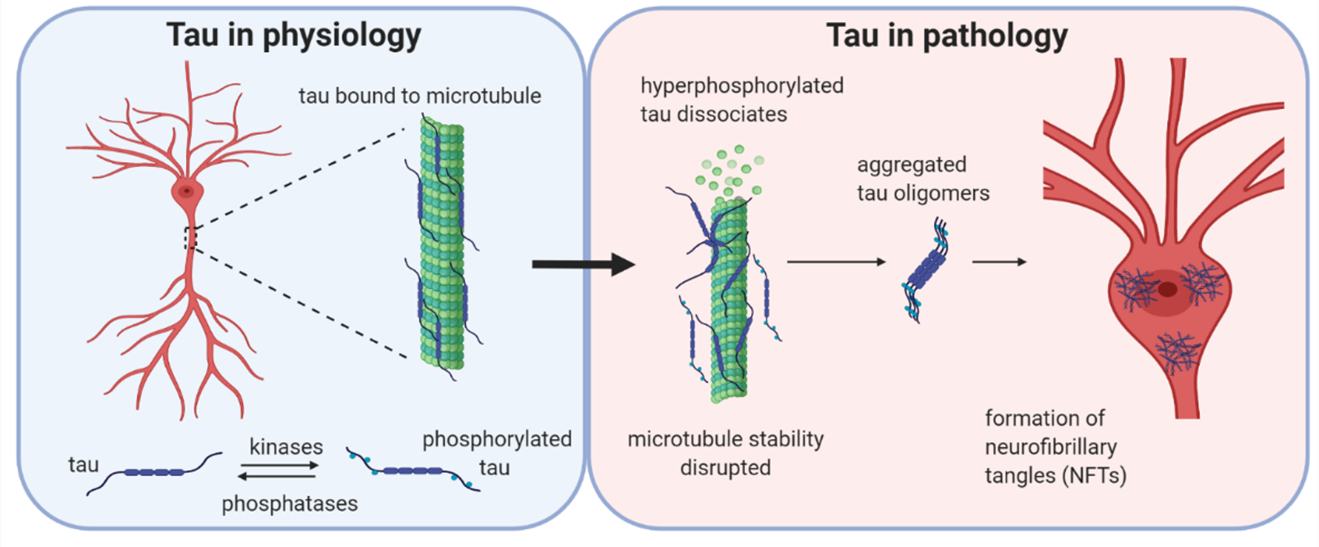
Under physiological conditions, tau is bound to microtubules and the stability is maintained by the balanced activities of kinases and phosphatases acting to regulate the binding of tau to the microtubules. If the balance is altered in favour of increased kinase activity, then tau can become hyperphosphorylated and detach from the microtubules. Initially, monomers are aggregation incompetent and have to undergo a conformational change using interactions via their hexapeptide motif or by disulphide bonding. Tau can then form aggregates, firstly small soluble oligomers which are β-sheet-rich and are thought to be the smallest toxic species. Subsequently, these species aggregate further into protofibrils, fibrils and finally into neurofibrillary tangles (NFTs).
Electrophysiological studies have revealed several pathological mechanisms for the actions of full length (FL) oTau, including alterations in neuronal excitability and changes in short- and long-term synaptic plasticity (8–11). Viral introduction and transgenic models are commonly used to explore the roles of oTau in pathology (5–7). While these methods generate valuable data, they provide little information on the concentration and structural conformations of oTau that are responsible for the neurotoxic effects. For other approaches, such as the extracellular application of oTau to tissue or cell cultures (12–14), cellular uptake may be the limiting step, reducing the observed toxicity.
To address these limitations, we have used whole-cell patch-clamp recording to introduce oTau into single neurons and has enabled specific targeting to either pre- or post-synaptic cells (8,15). This approach only requires small amounts of oligomers, each cell acts as its own control and electrophysiological alterations can be observed in real time. Furthermore, direct comparisons can be made between different concentrations and structural conformations of the oTau introduced. Using this approach, we have previously demonstrated that full-length (FL, prepared from recombinant full-length human tau 1-441) oTau (44-444 nM) modifies the excitability (as measured by a change in firing rate), input resistance and action potential waveform of CA1 pyramidal neurons.
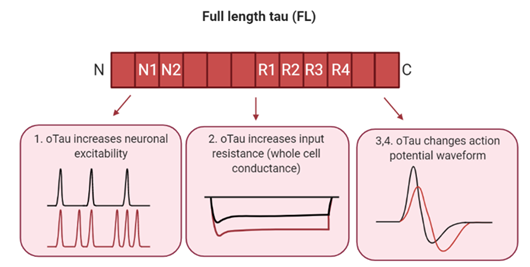
Introduction of recombinantly produced full length human tau (1-441) in oligomeric form (FL-oTau) into single neurons increases neuronal excitability (measured by a change in the rate of firing), increases input resistance (a given current producing a larger voltage response, indicative of a reduction in whole cell conductance) and alters action potential dynamics.
We hypothesized that using specific truncations of the tau molecule, we would be able to dissect apart these effects, which would help in identifying the underlying mechanisms. We therefore created truncated versions of tau, informed by a physiologically relevant truncation at amino acid 123 that was recently discovered in AD patients. We used oligomers formed from tau lacking the N-terminal region (aa 124-441; CFRAG) and soluble aggregates of the non-oligomer forming N-terminal fragment (aa 1-123; NFRAG).
We found that CFRAG-oTau did not replicate the effects on action potential kinetics or input resistance that were observed with FL-oTau but did still increase firing rate. We then showed that this firing rate increase is mediated by a reduction in the minimum current required to elicit an action potential, which in turn results from a reduction in the spike initiation threshold. Thus, the neurons become more excitable.
Conversely, we found that NFRAG-Tau aggregates changed the action potential height and width without increasing input resistance or excitability. Surprisingly, NFRAG monomers produced comparable changes to action potentials. Thus, the effects of NFRAG are likely to be unrelated to its quaternary structure and instead maybe associated with a specific sequence within the NFRAG tau.
Given the nature of the changes in action potential waveform and excitability, we hypothesised that tau might be interacting with voltage-gated sodium channels and altering their function. We adapted a published protocol to allow us to record voltage gated sodium currents in acute slices. We found that FL-oTau had three effects on sodium channel currents: Firstly, the half activation was reduced, reflecting the increase in neuronal excitability, consistent with the effects of CFRAG-oTau. Secondly, the rate of activation was reduced underlying the slower action potential rise, consistent with the effects of NFRAG-tau. Finally, FL-oTau reduced the maximum sodium conductance by ~ two thirds. To investigate the effects this would have on the AP, we implemented a simplified action potential model (16). Reducing the maximum sodium conductance had effects that replicated our data (reduced action potential amplitude and change to threshold).
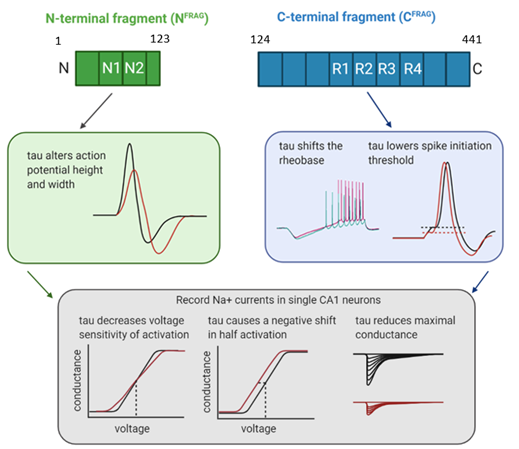
By truncating tau, we were able to dissect apart the effects on the action potential waveform and neuronal excitability. Using a combined experimental and mathematical modelling approach, we were then able to demonstrate that FL-oTau alters voltage gated sodium channel conductance and that this change could feasibly underpin both the changes to excitability and action potential dynamics.
This simple, yet highly effective technique of introducing structurally defined aggregated proteins into single neurons has allowed us to study the actions of tau with unparalleled levels of detail and provides a unique opportunity to understand the underlying pathology for tauopathies. By truncating the tau molecule, we have probed the mechanisms that underlie tau dysfunction, and this increased understanding of tau’s pathological actions will build towards developing future tau-targeting therapies.
For more details, see Hill et al., Communications Biology, 2021. https://www.nature.com/articles/s42003-021-02791-x
Follow the Topic
-
Communications Biology
An open access journal from Nature Portfolio publishing high-quality research, reviews and commentary in all areas of the biological sciences, representing significant advances and bringing new biological insight to a specialized area of research.

Related Collections
With Collections, you can get published faster and increase your visibility.
Healthy Aging
Publishing Model: Open Access
Deadline: Dec 31, 2026
Cell death and inflammatory signalling
Publishing Model: Hybrid
Deadline: Oct 28, 2026



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in