The effect of inhibition of receptor tyrosine kinase AXL on DNA damage response in ovarian cancer
Published in Cancer
Ovarian cancer (OC) patients who developed resistance to platinum-based chemotherapy generally respond poorly to the second-line treatment and require novel therapeutic options. AXL, a receptor tyrosine kinase, has been found to overexpress in the tumors of this group of patients. In a previous study, our research group identified AXL to be highly expressed in the mesenchymal (Mes)-subtype ovarian tumor tissue and cancer cell lines. By treating various OC cells with the first-in-class AXL inhibitor R428, the Mes-subtype showed selective sensitivity towards AXL inhibition. This thus makes AXL an appealing candidate as a new therapeutic target in OC.
Emerging studies have also brough to light a novel role of AXL in regulating DNA damage response (DDR). Since DDR is an apparent therapeutic target pathway with many assets being developed clinically, in this study, we were curious whether AXL inhibition would elicit DDR in the Mes-subtype OC and increase the sensitivity towards DDR inhibition as well.
Is there rationale to combine AXL and DDR inhibitors to treat OC?
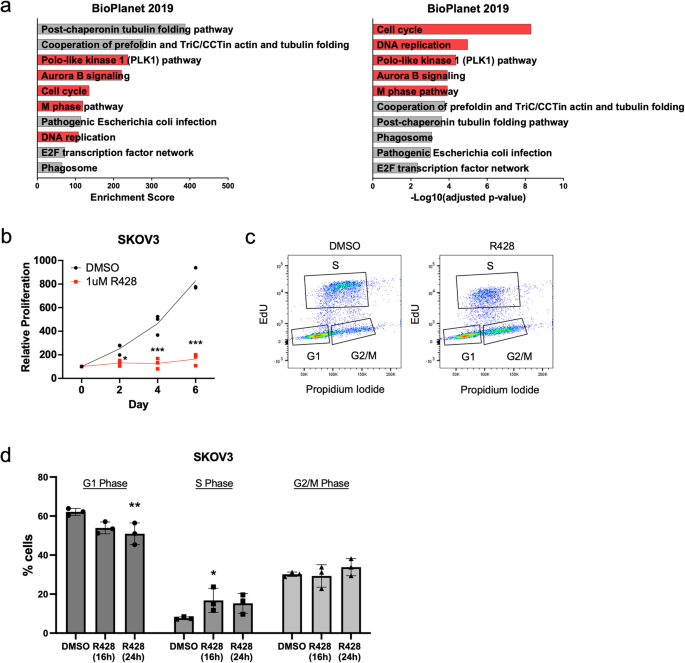
We first treated a Mes-subtype OC cell line expressing high AXL with R428 and explored the global transcriptomic changes. Among differentially expressed genes and pathways, genes associated with cell cycle, mitosis, and DNA replication were downregulated upon AXL inhibition. We confirmed that the decrease in proliferation was due to a G2/M phase arrest caused by replication stress. Markers for single-stranded DNA (ssDNA) and replication stress (γH2AX, pRPA32) were upregulated and many DDR signaling molecules such as ATM and ATR were activated. These results proved the point that AXL inhibition would indeed induce DDR.
We then explored the effects of a combination treatment using the AXL inhibitor R428 with an ATR inhibitor BAY1895344 in OC. ATR is a serine/threonine-kinase that senses the presence of ssDNA at stalled replication forks and activates DNA damage checkpoint leading to cell cycle arrest. The combination of ATR and AXL inhibition indeed induced DNA damage to catastrophic levels, leading to additive effects in cell death. By using both the in vitro assays and in ovo chick chorioallantoic membrane (CAM) xenograft experiments, we were able to show that AXL inhibition rendered OC cells more sensitive to ATR inhibition by further increasing DNA damage.
Now the question is: how does AXL mediate DDR in OC?
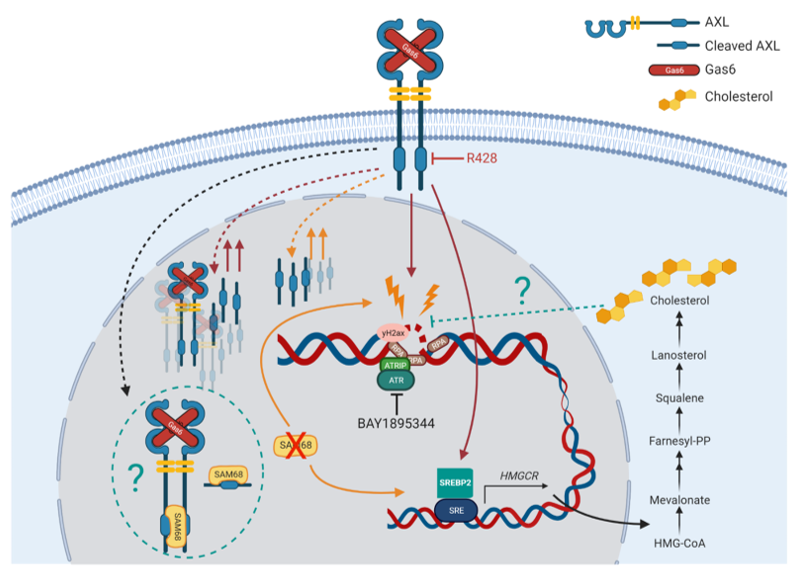
Like the EGFR family, AXL is known to exist as a nuclear form. After AXL inhibition in OC cells, both the full-length and cleaved AXL were detectable in the nuclear extract. This piece of evidence provides us with good basis to explore the nuclear role of AXL. We started by exploring potential binding partners of AXL inside the nucleus. By using stable isotope labelling by amino acids in cell culture (SILAC) co-immunoprecipitation (Co-IP) mass spectrometry, we identified a novel nuclear binding partner of AXL, Src associated in mitosis of 68 kDa (SAM68/KHDRBS1). SAM68 is a RNA-binding protein that plays a variety of roles in cellular processes including RNA stability and nuclear export. Emerging evidence shows that SAM68 has critical functions in DDR. This discovery thus formed a strong rationale to elucidate the role of AXL-SAM68 interaction in DNA damage.
What was intriguing was that AXL inhibition and the loss of SAM68 in OC cells resulted in similar phenotypes in DDR. The loss of SAM68 could also sensitize OC cells to the combination treatment of AXL and ATR inhibitors. However, the presence of nuclear AXL and the DDR initiated by AXL inhibition was not affected by the loss of SAM68. Our finding suggested that despite AXL and SAM could be protein binding partners, there might exist parallel and independent roles of AXL and SAM68 in the regulation of DDR.
Since ALX and SAM68 have parallel roles in the regulation of DDR, can we explain the shared DDR phenotypes via one unifying mechanism?
We then decided to explore the overlapping functions or pathways that could be regulated by AXL and SAM68 and would link to the shared DDR phenotype. Transcriptomic analysis performed on AXL and SAM68 deficient cells showed that AXL- and SAM68-deficiency or AXL inhibition induced elevated levels of cholesterol and upregulated genes in the cholesterol biosynthesis pathway. This were validated in several experiments including the expression levels of genes and proteins related to the cholesterol biosynthesis pathway (SREBF2, ACAT2, HMGCS1, HMGCR, FDFT1), and the cholesterol levels demonstrated through filipin III staining, cholesterol quantification by Amplex™ Red Cholesterol Assay, and liquid chromatography–mass spectrometry (LC–MS).
These findings thus prompted us to investigate whether cholesterol biosynthesis would be the answer to the mechanism of DDR induced by AXL inhibition.
Cholesterol might have a protective role in shielding OC cells against DNA damage induced by AXL inhibition or SAM-deficiency.
We were able to show that supplementation of cholesterol could attenuate the decrease in proliferation caused by AXL inhibition, suggesting that the increase in cholesterol biosynthesis upon AXL inhibition may be a survival mechanism that counteracts AXL inhibition. It is therefore plausible that cholesterol may prevent or decrease DNA damage induced by AXL inhibition.
We knocked down the cholesterol biosynthesis genes SREBF2 or HMGCR1 to block downstream cholesterol biosynthesis and to lower the intracellular cholesterol levels. These cells demonstrated increased DDR and AXL inhibition could further enhance DDR in these cells. Importantly, supplementation of cholesterol delayed the increase in γH2AX expression induced by AXL inhibition in these cells, albeit not to the levels of the control cells.
Could "skinny" OC cells be more vulnerable to therapeutic interventions?
Our data clearly suggested that the amount of intracellular cholesterol might determine how OC cells mitigate DNA damage triggered by treatment such as AXL inhibition. One open question would thus be: is cholesterol biosynthesis the escape mechanism for treatment resistance in OC cells?
If this is true, we might want to hope for some skinny privilege for OC treatment after all.
Follow the Topic
-
Communications Biology
An open access journal from Nature Portfolio publishing high-quality research, reviews and commentary in all areas of the biological sciences, representing significant advances and bringing new biological insight to a specialized area of research.

Related Collections
With Collections, you can get published faster and increase your visibility.
Artificial Intelligence Methodology in Structural Biology
Publishing Model: Hybrid
Deadline: Nov 30, 2026
Healthy Aging
Publishing Model: Open Access
Deadline: Dec 31, 2026




Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in