The Insidious Lymphodepletive Effect of Myeloproliferative Neoplasms
Published in Cancer and Immunology
Explore the Research
JAK2V617F impairs lymphoid differentiation in myeloproliferative neoplasms - Leukemia
Leukemia - JAK2V617F impairs lymphoid differentiation in myeloproliferative neoplasms
It is well recognized that patients with myeloproliferative neoplasms (MPNs) risk abnormal blood clots due to the overproduction of myeloid cells like red blood cells, platelets and neutrophils. However, patients with MPNs are also prone to serious infections [1,2] and second cancers [3–5] that evoke immune system impairment more than myeloid proliferation. Markers of immune dysfunction like lymphopenia and elevated neutrophil-to-lymphocyte ratios, rather than just abnormal myeloid cell counts, predict shortened patient survival [6,7]. What drives this apparent immune dysfunction in MPNs is not known.
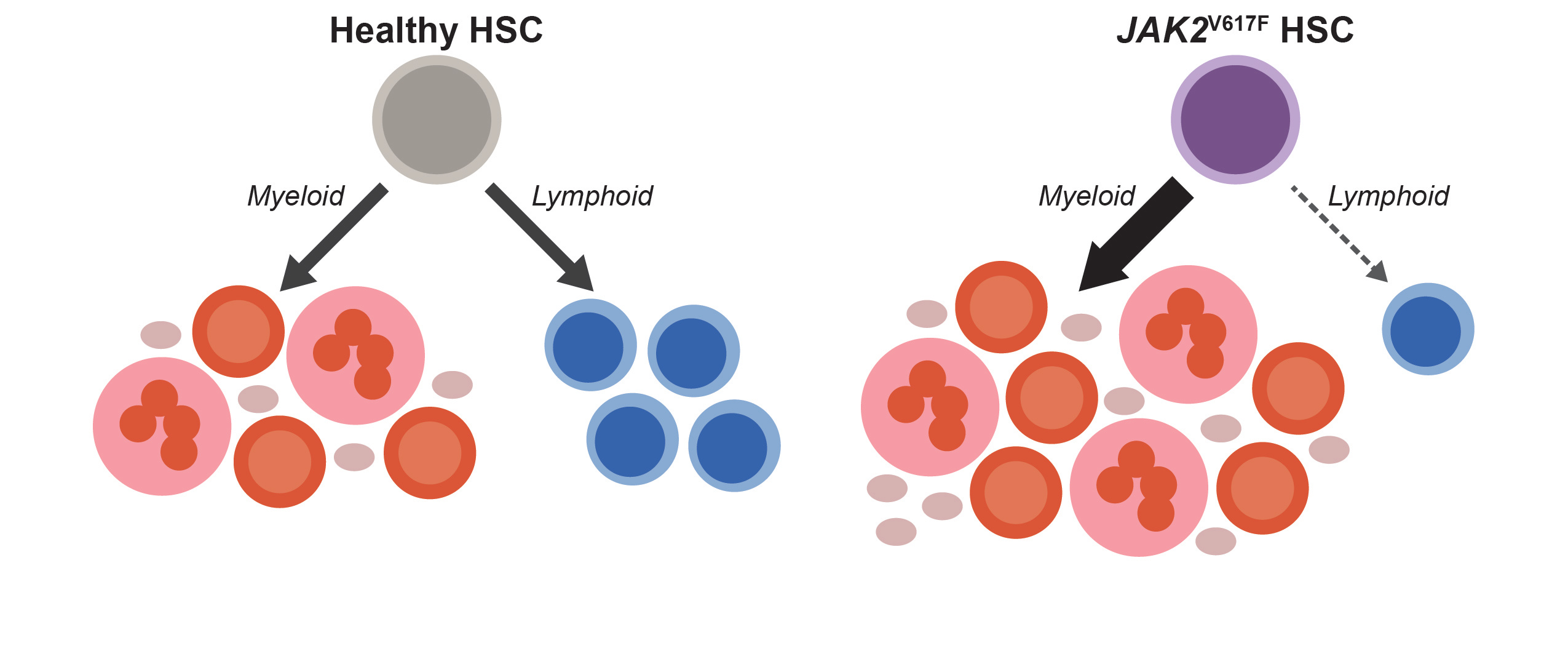
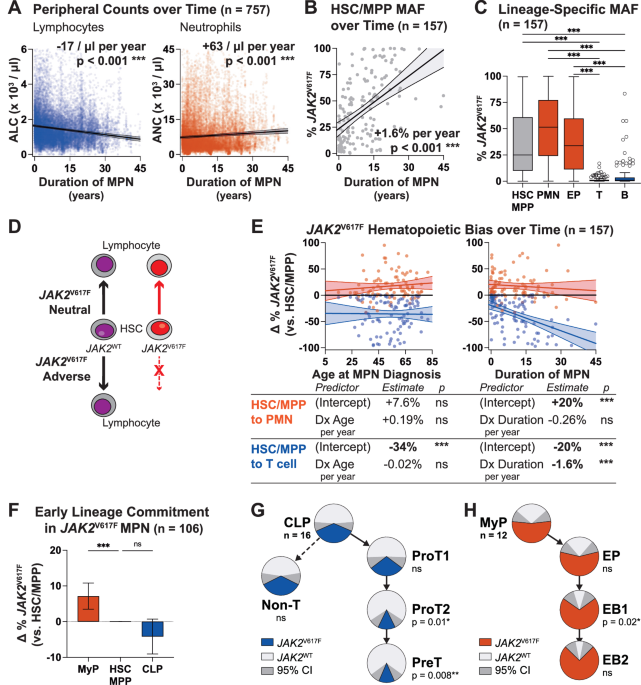
When we started thinking about the immune system in MPNs, a longstanding paradox caught our attention: although MPNs originate from hematopoietic stem cells (HSCs) that acquire a driver mutation (most frequently JAK2V617F), and HSCs normally give rise to both myeloid and lymphoid cells, JAK2V617F is almost never detected in lymphocytes [8–10]. Since most patients are diagnosed with MPNs after their fifth decade of life, while lymphocyte production is most active much earlier, one may assume that in most patients, JAK2V617F simply occurs after most lymphocytes have already been produced. However, we found that this assumption had never been tested. If it were true, we would expect that mutated lymphocytes would be more easily detected in patients who were diagnosed at a young age, and in those who had their MPNs for a long time. We longitudinally tracked JAK2V617F in lymphocytes from over 150 patients at our MPN Center and did not find this to be the case. In fact, even though patients accumulated more JAK2V617F HSCs over time, the mutation remained largely undetectable in lymphocytes. This finding suggests that even as JAK2V617F HSCs dominate myeloid cell production, they struggle to generate lymphocytes, as shown in the schematic below.

To test this possibility, we assessed lymphoid development from patient-derived HSCs and found that the JAK2V617F cells were depleted during T cell differentiation in vitro. We found through analysis of over 20,000 complete blood counts from our MPN Center that the longer patients had JAK2V617F MPN, the more their lymphocyte counts dropped. This relationship held regardless of patient age or therapy. Therefore, our patient data implicated insufficient lymphocyte production with advancing disease as a culprit for immune dysfunction in JAK2V617F MPNs.
Definitive support for these observations came from unexpected sources. One was a chimeric mouse model we had generated to evaluate hematopoiesis in early MPNs. We mixed small doses of HSCs bearing Jak2V617F (the mouse ortholog of human JAK2V617F) into the bone marrow of these mice to study how MPN HSCs establish themselves to initiate disease. Although our initial focus was on myeloid cells, we soon made a fortuitous discovery: the mutant HSCs failed to generate mature lymphocytes, just as we had observed in patients. This result found us just as we were completing our studies of T cell differentiation in humans, so we dug deeper into the mouse model. We found that Jak2V617F HSCs fail to complete T and B lymphoid development programs in these mice, and in the case of T cell development, the block occurred at the same stage of differentiation as we had observed from human cells.
While we were working through these findings, we had another stroke of luck. During a casual email exchange with our colleague in Bern, Dr. Tata Nageswara Rao, we found out that his lab was also studying lymphocyte production in MPNs, but with a different approach. His team had been using a transgenic mouse model expressing high levels of JAK2V617F in the HSCs. They found a very strong block in lymphoid development in these transgenic mice, which (in contrast to our chimeric mice) developed lymphopenia. They had also analyzed gene expression in their model to demonstrate that JAK2V617F was suppressing the Notch signaling pathway, which is critical to lymphocyte development. This corroborated an incidental observation we had made previously, that treating healthy human HSCs with a Notch inhibitor causes them to behave like JAK2V617F HSCs during T cell differentiation. Through our collaboration, we were able to connect multiple complementary findings to add depth to the understanding of how JAK2V617F disrupts lymphocyte development. A clear narrative emerged: JAK2V617F consistently interrupts lymphocyte production in MPNs by attenuating key lymphoid development signals, with a stronger effect in settings where JAK2V617F HSCs dominate, as in patients with advanced MPNs or the transgenic mice.
We’ve come to understand that, over time, defective lymphocyte production plays a central role in the immune deficiencies seen in MPNs. As JAK2V617F HSCs progressively dominate MPN hematopoiesis, lymphocyte production increasingly depends on the shrinking pool of normal HSCs. This strain ultimately leads to lymphopenia, thus compromising the foundation of adaptive immunity in patients with MPNs. Stresses related to maintaining lymphocyte production from this dwindling pool of normal HSCs may also explain the increased occurrence of lymphomas in patients with JAK2V617F MPNs, which, interestingly, almost always originate from cells without the JAK2V617F mutation [11].
Motivated by a paradox and propelled by unexpected findings and a fruitful collaboration, we discovered that MPNs are not just “myeloproliferative” but are also profoundly “lymphodepletive.” This dual nature of JAK2V617F-driven hematopoiesis provides a clear mechanistic explanation for the adverse clinical outcomes linked to lymphopenia in MPNs. This deeper understanding of MPN hematopoiesis highlights the importance of restoring the normal balance between myeloid and lymphoid production. Therapies that counteract JAK2V617F during both myeloid and lymphoid development to rebalance MPN hematopoiesis could improve immune function in patients. Looking forward, these insights point to promising therapeutic strategies for managing MPNs and improving patient outcomes.
References:
[1] Crodel CC, Jentsch-Ullrich K, Koschmieder S, Kämpfe D, Griesshammer M, Döhner K, et al. Frequency of infections in 948 MPN patients: a prospective multicenter patient-reported pilot study. Leukemia 2020;34:1949–53. https://doi.org/10.1038/s41375-020-0890-1.
[2] Landtblom AR, Andersson TM-L, Dickman PW, Smedby KE, Eloranta S, Batyrbekova N, et al. Risk of infections in patients with myeloproliferative neoplasms-a population-based cohort study of 8363 patients. Leukemia 2021;35:476–84. https://doi.org/10.1038/s41375-020-0909-7.
[3] Landtblom AR, Bower H, Andersson TM-L, Dickman PW, Samuelsson J, Björkholm M, et al. Second malignancies in patients with myeloproliferative neoplasms: a population-based cohort study of 9379 patients. Leukemia 2018;32:2203–10. https://doi.org/10.1038/s41375-018-0027-y.
[4] Barbui T, Ghirardi A, Masciulli A, Carobbio A, Palandri F, Vianelli N, et al. Second cancer in Philadelphia negative myeloproliferative neoplasms (MPN-K). A nested case-control study. Leukemia 2019;33:1996–2005. https://doi.org/10.1038/s41375-019-0487-8.
[5] Zhang Y, Han Y, Teng G, Du C, Gao S, Yuan W, et al. Incidence and risk factors for second malignancies among patients with myeloproliferative neoplasms. Cancer Med 2023;12:9236–46. https://doi.org/10.1002/cam4.5666.
[6] Tefferi A, Loscocco GG, Farrukh F, Szuber N, Mannelli F, Pardanani A, et al. A globally applicable “triple A” risk model for essential thrombocythemia based on Age, Absolute neutrophil count, and Absolute lymphocyte count. Am J Hematol 2023;98:1829–37. https://doi.org/10.1002/ajh.27079.
[7] Larsen MK, Skov V, Kjær L, Eickhardt-Dalbøge CS, Knudsen TA, Kristiansen MH, et al. Neutrophil-to-lymphocyte ratio and all-cause mortality with and without myeloproliferative neoplasms—a Danish longitudinal study. Blood Cancer J 2024;14:28. https://doi.org/10.1038/s41408-024-00994-z.
[8] Jamieson CHM, Gotlib J, Durocher JA, Chao MP, Mariappan MR, Lay M, et al. The JAK2 V617F mutation occurs in hematopoietic stem cells in polycythemia vera and predisposes toward erythroid differentiation. Proc Natl Acad Sci U S A 2006;103:6224–9. https://doi.org/10.1073/pnas.0601462103.
[9] Butcher CM, Hutton JF, Hahn U, To LB, Bardy P, Lewis I, et al. Cellular origin and lineage specificity of the JAK2V617F allele in polycythemia vera. Blood 2007;109:386–7. https://doi.org/10.1182/blood-2006-07-036426.
[10] Abu-Zeinah G, Di Giandomenico S, Choi D, Cruz T, Erdos K, Taylor E, et al. Hematopoietic fitness of JAK2V617F myeloproliferative neoplasms is linked to clinical outcome. Blood Adv 2022;6:5477–81. https://doi.org/10.1182/bloodadvances.2022007128.
[11] Swierczek S, Nausova J, Jelinek J, Liu E, Roda P, Kucerova J, et al. Concomitant JAK2 V617F-positive polycythemia vera and B-cell chronic lymphocytic leukemia in three patients originating from two separate hematopoietic stem cells. Am J Hematol 2013;88:157–8. https://doi.org/10.1002/ajh.23362.
Follow the Topic
-
Leukemia
This journal publishes high quality, peer reviewed research that covers all aspects of the research and treatment of leukemia and allied diseases. Topics of interest include oncogenes, growth factors, stem cells, leukemia genomics, cell cycle, signal transduction and molecular targets for therapy.





Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in