The quest to organize the prokaryotic virosphere
Published in Bioengineering & Biotechnology
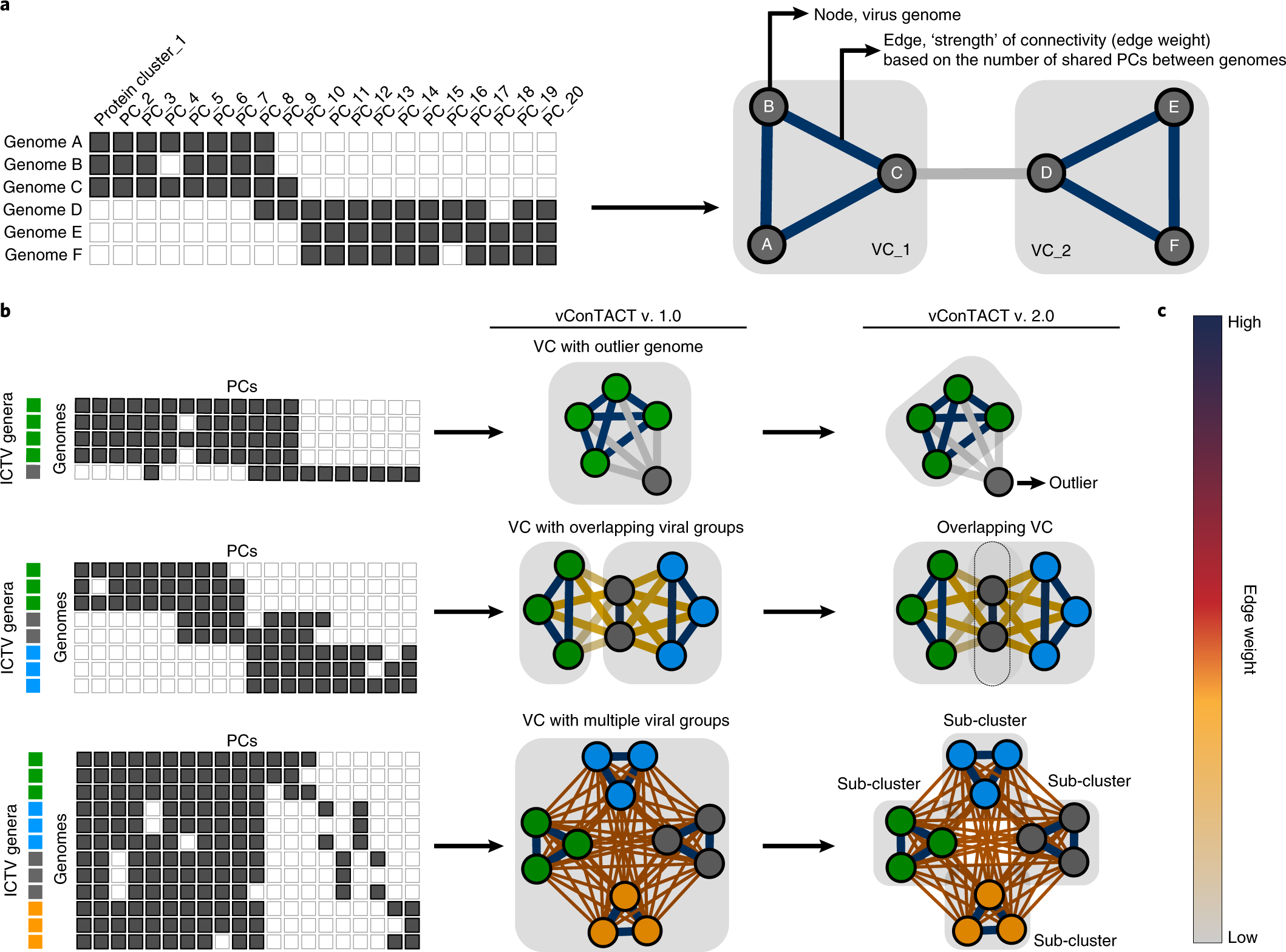
The abundance of bacterial viruses (‘phages’) on Earth is inconceivably high – rivalling the number of observable stars in the night sky, so how do we organize this immense viral space in a robust, taxonomic classification system? One would immediately think that perhaps using a virus gene present in every virus genome would do the trick, like the cherished 16S rRNA gene for microbes—alas no such ‘golden’ gene exists, likely because virus have evolved separately and do not have a single universal ancestor virus that gave rise to what we see today. So what do we use? Away from the classic virus taxonomist toolkit, we had to forget about virion morphology to guide us, since most of the viruses discovered in modern studies are only known by (partial or complete) genomic sequence. Could the genome alone really help us organize the virosphere? And if structure in viral sequence space did emerge, would these groupings be similar to those created by decades of manually created virus groups by dedicated virus taxonomists?
The idea of a ‘universal’ classification framework can be thought as theoretically flawed from the get-go: if there is no universality in virus genomes, how could a universal classification system—which implies consistency —could ever be devised? We faced two major challenges in attempting to go forward with this apparent irrationality. The first is scale— with the constant deluge of new uncultivated virus genomes that metagenomics offered us, the classical way to classify a virus using morphology (which we don’t have anyway) and single-gene dendrograms would be impossible to do manually. To put that into perspective, it took 50 years to achieve the current classification of ~800 phages, or 16 phages per year on average. At this rate, to classify say 100,000 uncultivated phages, it would take us over 6000 years to complete, so we had to find a better way!
The second major hurdle is for the framework to consider variations in gene flow/recombination between phage genomes. The paradigm is that gene flow in phages is so great that it ameliorates taxonomic boundaries, but what if instead it was a cohesive evolutionary force as found for bacteria, archaea and recently deeply sequenced phage groups (e.g., cyanophages and Mycobacterium phages). Phages likely have variable gene flow rates to enable myriad evolutionary strategies to succeed. If too much gene flow is occurring then our gene sharing networks would be obfuscated and inconsistent with known taxonomy, but if gene flow were constrained to like relatives then perhaps the structure we (hoped!) to observe might be consistent with accepted taxonomy. We needed ideas outside of phylogenetic trees and ‘universal’ shared protein cutoffs. These had not saved the day.
In 2008, a revolutionary approach to phage classification appeared in the literature1, which described the application of networks to deal with rampant gene flow between phages. It worked very well, reaching >90% accuracy in mirroring the official phage classification at the time. However, there remained difficulties for resolving some phage groups that had the highest recombination rates. The most rampant ones—of them the Mycobacterium phages. Some improvement was made by several groups that dived deeper into the network strategy to phage classification, but none were broadly implemented nor thought to be a viable alternative to classical phylogenetic analyses (Figure 1).

Nearly a decade later, in 2017, led by postdoctoral researcher Dr. Ben Bolduc (OSU) we re-evaluated the network approach on a much larger reference dataset of viruses that had grown significantly since the initial network-based efforts1 work, and developed an optimized tool, vConTACT12, as a more grounded, publicly-available application that would broaden its usage. Although an improvement, vConTACT1 lacked features that would enable automated taxonomic classification in part because confidence measures per virus grouping predictions were not available, and some regions of viral genomic space could not be accurately grouped due to high mosaicism for certain virus groups like—you guessed it— Mycobacterium phages.
For the development of the next vConTACT version, Dr. Ho Bin Jang (OSU) identified the underlying causes of erroneous grouping for high gene flow virus groups, which called for a change in the core network algorithm. Additional grouping methods were added, along with multiple measures of confidence for each predicted virus group. Collectively, these changes led to a global >96% accuracy in genus-level predictions for the entire viral RefSeq. Finally, rampant mosaic genomes could be resolved, and the network could provide guidance for the decision-making processes that have led to ratified taxonomic proposals.
A lingering question remained: granted, we could now accurately predict virus groupings, but would our predictions still hold true if we were to add hundreds or even thousands of new genomes to the network? It could go in two ways: either the virus space is a giant interconnected web of shared genes being shuffled around and as we add more, all viruses will end up being reshuffled into different groups until we reach saturation and all lump into one group; OR there are discreet, biological groups that remain separate regardless of new data—the idea of a structure—in viral space. The opportunity was there to test this. For some groups of viruses, such as the cyanophages (marine viruses) we knew this structure existed, founded upon niche-driven speciation. But what about the entire known prokaryotic virosphere? At the same time, we needed to address one additional challenge of our classification framework: will it scale to thousands of genomes?
To hit two phages with one stone, we used the global ocean virome dataset—GOV— a collection of nearly 16,000 uncultivated virus genomes collected throughout the world’s oceans. When it came to scaling up, vConTACT2 performed flawlessly, although generating a network image of that many viruses could be argued as being the most compute intensive analysis step! We could now answer how the virosphere was organized: instead of a chaotic interconnected web, we saw that there was indeed structure, and that the original reference network barely changed with the addition of thousands of marine viruses.
Hopefully, we now have a tool to organize, in the future, the entire virosphere, and look at viruses at the big data scale to reveal us global patterns of diversity and gene-exchange of the most ubiquitous organism on the planet.
We had reached our goals, but the path was arduous at every turn. This work was highly collaborative, and is the result of extensive contributions from dozens of individuals across disciplines. Members of the International Committee on Taxonomy of Viruses (ICTV) provided guidance for taxonomic rigor and with manuscript revisions, and included Andrew Kropinski (U. Guelph), Evelien Adriaenssens (Quadram Institute), Rodney Brister (NCBI), and Rob Lavigne (KU Leuven). This work was also possible by the contributions of Jens Kuhn (NIH), Simon Roux (JGI), Mart Krupovic (Institut Pasteur), Dann Turner (UWE Bristol) and Wesley You (OSU) for their guidance, manuscript writing and bioinformatics support. We are grateful for the support of the CyVerse infrastructure staff, whose collaboration enabled the deployment of vConTACT2 on their platform. We wish to thank the people who reviewed our work for taking the time to provide us with essential feedback and suggestions to improve our work, as well as to the people who tested vConTACT2, particularly Consuelo Gazitua, Gary Trubl, Ann Gregory, and Dean Vik. Lastly, from Ho Bin, Ben Bolduc and I, we would like to personally acknowledge our mentor, Matt Sullivan, for his continuous support, and whose passion and leadership has enabled us to produce this work.
References
1. Lima-Mendez, G., Van Helden, J., Toussaint, A. & Leplae, R. Reticulate representation of evolutionary and functional relationships between phage genomes. Mol. Biol. Evol. 25, 762–777 (2008).
2. Bolduc, B. et al. vConTACT: an iVirus tool to classify double-stranded DNA viruses that infect Archaea and Bacteria. PeerJ 5, e3243 (2017).
Follow the Topic
-
Nature Biotechnology
A monthly journal covering the science and business of biotechnology, with new concepts in technology/methodology of relevance to the biological, biomedical, agricultural and environmental sciences.



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in