The role of Cyanide in origins of protometabolism on early earth
Published in Chemistry
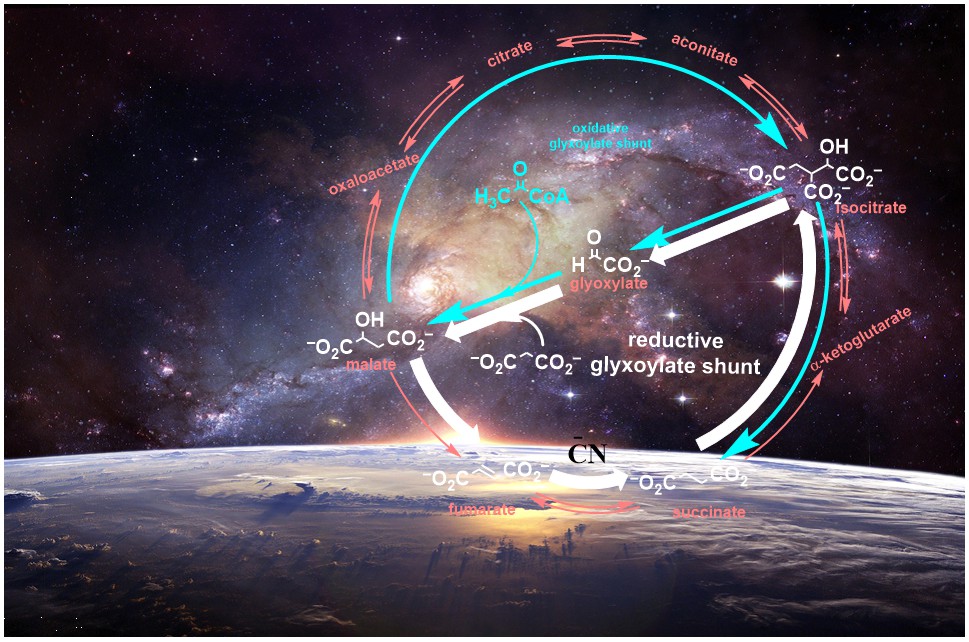

Extant biology uses a complex set of biochemical transformations (metabolism) which are governed by highly specific enzymes.1 Many of these metabolic reactions either take up CO2 to build up bigger organic molecules (anabolism) or release CO2 via breakdown of complex molecules (catabolism) to smaller ones. But how such reactions (or similar reactions) could have been developed on the prebiotic earth is still a question that is of interest to the prebiotic community.2,3 Skepticism has been expressed that proto-metabolic pathways on early earth work in an anabolic way to make organic molecules by fixation of CO2. Therefore, two ancient CO2 fixation cycles have been pointed out as being potentially prebiotically plausible: one of them is reverse, reductive citric acid (Tri Carboxylic Acid, rTCA or reverse Krebs) cycle and the other is AcCoA pathway (also known as the Wood-Ljungdahl pathway).3 So far, most of the protometabolism approaches have focused mainly on the reductive citric acid cycle (Fig. 1, inner cycle). In this direction, efforts have been made to reductively fix CO2 with the help of metal catalysis to replicate r-TCA cycle, however, with little or no success.3
Considering all these difficulties, we thought of another feedstock molecules which are available of early earth and has a potential to react with other prebiotic molecules. Hydrogen cyanide (HCN) is such a source molecule, and its reaction potential has been well demonstrated in the prebiotic chemistry.4,5 Additionally, cyanides/nitriles are already at a reduced state when compared to carbon dioxide (cyanide is the nitrogen containing equivalent of carbon monoxide). Thus, accessing molecules at a ‘reduced’ state from nitriles would be relatively straightforward and easier (compared to starting from CO2 whose reduction under prebiotic conditions is quite difficult to accomplish). An example of such a reduction (glyoxylate to tartrate) has been demonstrated in presence of cyanide recently by Krishnamurthy lab.6
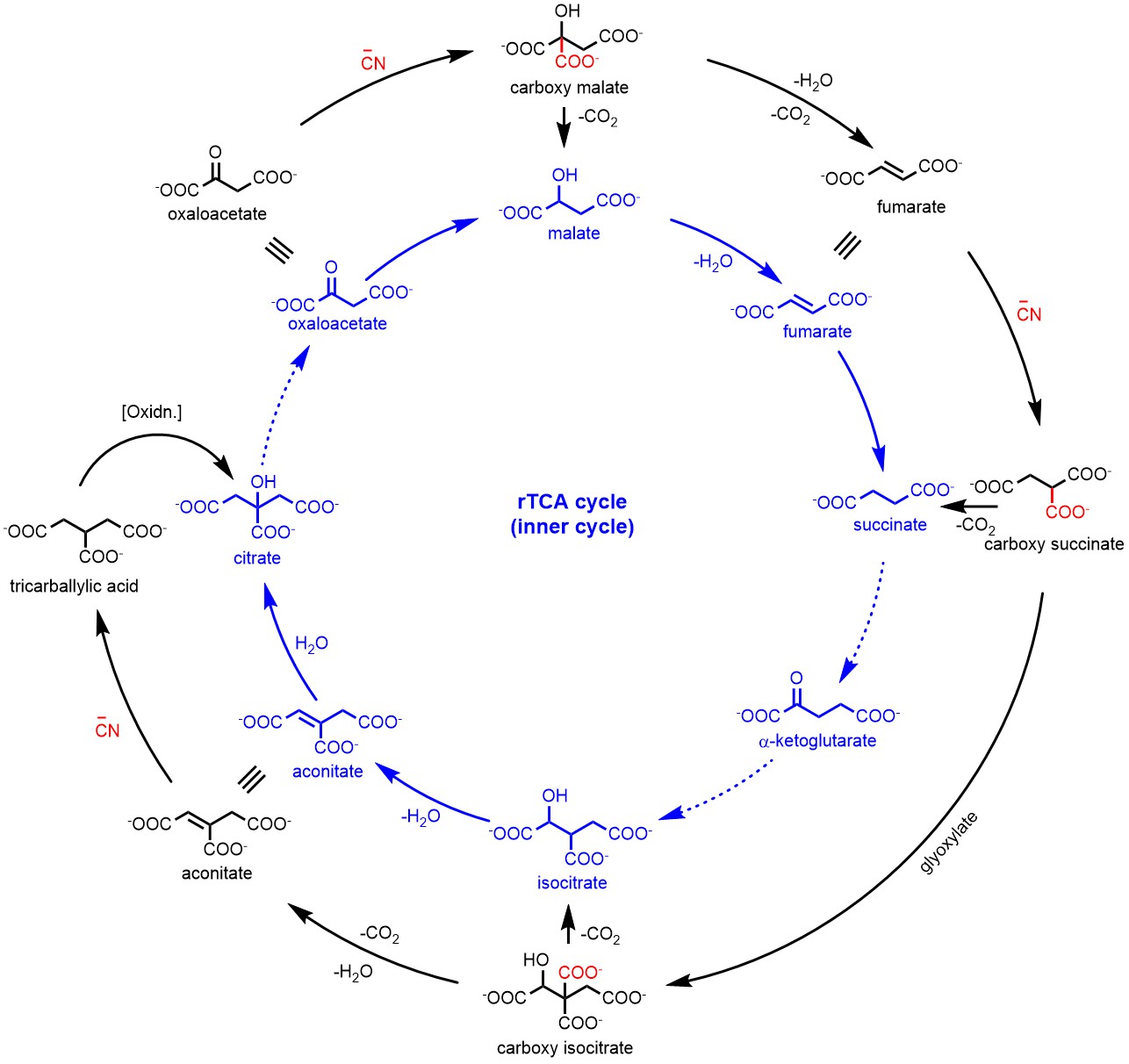
Figure 1: The cyanide-mediated transformations (outer black cycle) of molecules in the r-TCA cycle (inner blue cycle). Solid arrows represent the transformations achieved in this study while dashed arrows represent the bypassed or unachieved transformations.
Motivated by these thoughts, we investigated the reaction of cyanide with oxaloacetate under mildly acidic conditions. To our delight, the formation of carboxymalate was observed in almost quantitative yield which was easily decarboxylated to malate. Interestingly, this transformation mimicked a step of rTCA cycle, however, without the need of any metal or enzymatic catalysis. We explored the dehydration of malate to fumarate and this subsequent step of rTCA cycle also worked efficiently in the presence of ammonium formate (hydrolysis product of HCN). With the success of these two steps, we were curious about the next transformations of rTCA cycle i.e. fumarate to succinate. The fumarate to succinate conversion is indeed a reduction; however, unlike the conversion of oxaloacetate to malate, it involves the reduction of double bond. We envisioned this conversion possible with the addition of cyanide to the conjugated double bond of fumarate, however, this reaction did not proceed at near neutral pH. Interestingly, based on a report about hydration of monoanionic fumarate to malate7 we investigated moderately acidic conditions, and observed that monoanionic fumarate was converted to succinate through carboxy succinate in the presence of cyanide in very good yields. The rTCA cycle involves the conversion of succinate to 2-ketoglutarate, a reductive carbonylation of succinate which is highly challenging without any catalysis. Interestingly, the glyoxylate shunt pathway, suggested as a plausible precursor to the modern oxidative TCA cycle, bypasses the α-ketoglutarate to produce the isocitrate from succinate.8 However, the conversion of succinate to isocitrate is also challenging without catalyst due to low acidity of the protons in succinate. However, carboxysuccinate, an intermediate formed in the reaction of cyanide with fumarate has an extra carboxylate group which increases the acidity of α-proton that can enable reaction with glyoxylate. Interestingly, when carboxy-succinate was reacted with glyoxylate, it produced the carboxy-isocitrate which upon decarboxylation afforded the isocitrate as a mixture of diastereomers. The formation of isocitrate directly from fumarate via carboxysuccinate (bypassing 2-ketoglutarate) was highly intriguing and prompted us to investigate the potential transformations of isocitric acid. When isocitrate was heated under aqueous conditions in presence of ZnCl2, aconitate was produced that in the presence of Zn2+ afforded citrate. Coincidentally , these non-enzymatic transformations seem to parallel the rTCA cycle. In this way, we have shown the potential of cyanide that enable most of the steps of the rTCA cycle without the involvement of enzymatic catalysis. The retro-Aldol reaction of citrate should produce oxaloacetate and acetate; but, the abiotic versions are inefficient to make the r-TCA cycle self-sustaining in a prebiotic context.9
However, with the involvement of cyanide, we have identified an alternative pathway (reverse glyoxylate pathway) when starting with malonate and glyoxylate which bypasses the formation of all of the keto acid intermediates of the rTCA cycle and avoids the difficulties of trying to make an abiotic version of the r-TCA cycle involving CO2 explicitly as a reactant. The simplicity of the reductive-glyoxylate reactions suggest that such a pathway can exist on the early Earth as a forerunner of today’s metabolism. To read more about the Reverse Glyoxylate Pathway please see the publication (Nat. Chem., 2022, 14, 170-178).10
In addition to enable several useful transformations, the cyanide chemistry presented here also alleviates a major concern that these very same abiotic processes would interfere by irreversibly reducing pyruvate and α-ketoglutarate to their corresponding α-hydroxy acids (figure 2).11
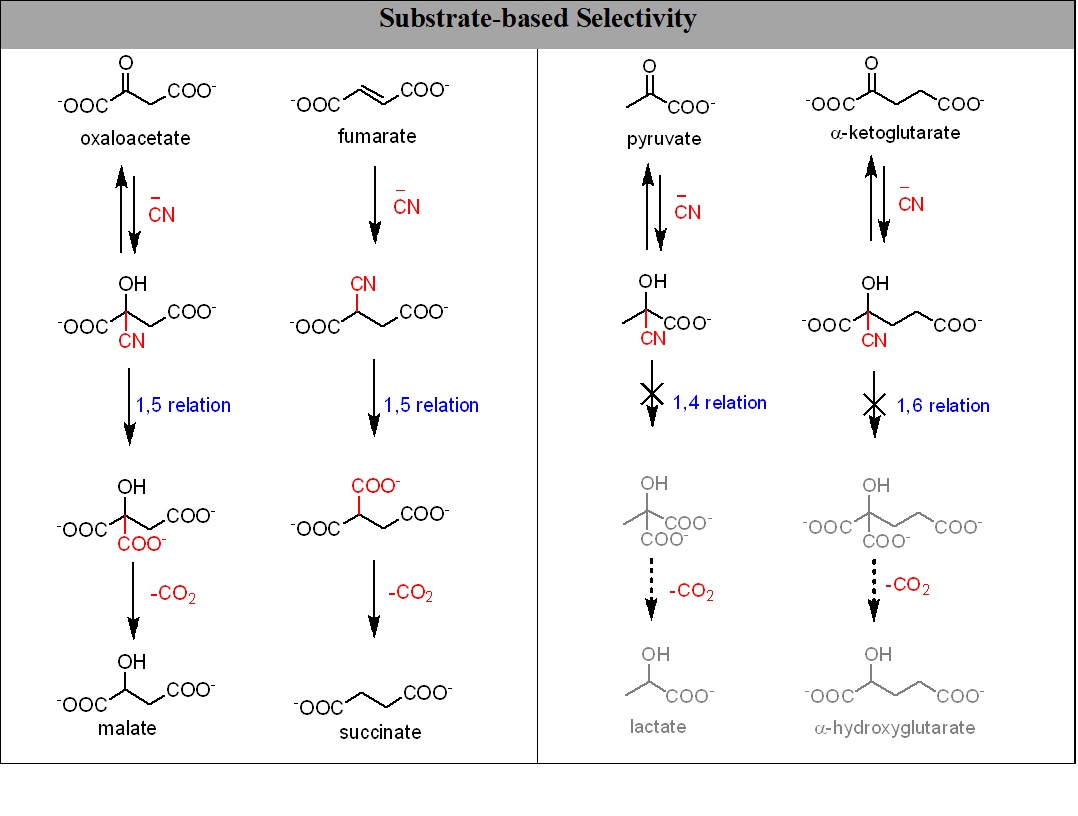
Figure 2: The cyanide-mediated substrate dependent selectivity in the reductive transformations.
In the contemporary rTCA cycle, oxaloacetate is reduced to malate while α-ketoglutarate and pyruvate are not reduced to their corresponding hydroxy acids. Such a selective reduction on the early earth has been elusive in the absence of sophisticated catalysis. However, the cyanide-based chemistry presented here nicely illustrates that such a selection criterion can come from the substrate only (while interacting with other small molecules). Figure 2 explicitly shows that in presence of cyanide oxaloacetate and fumarate are reduced to malate and succinate respectively while pyruvate and 2-ketoglutarate are not. All of the above stated molecules make the corresponding cyanohydrins in the presence of cyanide but for the hydrolysis of cyanohydrin the corresponding hydroxy acid, 1-5 anchimeric assistance is required which can be seen only in the case of oxaloacetate and fumarate.6 As a result of this ‘selection by substrate’, oxaloacetate and fumarate get reduced while pyruvate and 2-ketoglutarate do not.
In summary, in this study we have demonstrated that cyanide (a simple molecule) not only act as a mild and efficient reductant that enables abiotic transformations selectively in the r-TCA cycle that were once thought only to belong to biology and highly evolved enzymes, but also makes possible alternative abiotic pathways avoiding many difficult (abiotic) transformations.9 Such a change in the paradigm could enable a different way of thinking about the abiotic chemical roots of the origins of biochemistry in the context of origins of life on the early Earth.
References:
- McMurry, J. & Begley, T. The Organic Chemistry of Biological Pathways (Roberts, 2005).
- Morowitz, H. J., Kostelnik, J. D., Yang, J. & Cody, G. D. The origin of intermediary metabolism. Proc. Natl Acad. Sci. USA 97, 7704–7708 (2000).
- Muchowska, K. B., Varma, S. J. & Moran, J. Chem. Rev. 120, 7708–7744 (2020).
- Eschenmoser, A. On a hypothetical generational relationship between HCN and constituents of the reductive citric acid cycle. Chem. Biodivers. 4, 554–573 (2007).
- Yadav, M., Kumar, R. & Krishnamurthy, R. Chemistry of abiotic nucleotide synthesis. Chem. Rev. 120, 4766–4805 (2020).
- Yerabolu, J. R., Liotta, C. L. & Krishnamurthy, R. Anchimeric-assisted spontaneous hydrolysis of cyanohydrins under ambient conditions: implications for cyanide-initiated selective transformations. Chem. Eur. J. 23, 8756–8765 (2017).
- Bender, M. L. & Connors, K. A. A non-enzymatic olefinic hydration under neutral conditions: the kinetics and mechanism of the hydration of fumaric acid monoanion. J. Am. Chem. Soc. 84, 1980–1986 (1962).
- Zubay, G. The glyoxylate cycle, a possible evolutionary precursor of the TCA Cycle. Chemtracts 16, 783–788 (2003).
- Cody, G. D., Boctor, N. Z., Hazen, R. M., Brandes, J. A., Morowitz, H. J., Yoder, H. S., Jr. Geochemical roots of autotrophic carbon fixation: hydrothermal experiments in the system citric acid, H2O-(±FeS)−(±NiS). Cosmochim. Acta 65, 3557− 3576 (2001).
- Yadav, M., Pulletikurti, S., Yerabolu, J. R. & Krishnamurthy, R. Nat. Chem. 14, 170-178 (2022).
- Orgel, L. E. The implausibility of metabolic cycles on the prebiotic earth. PLoS Biol. 6, 0005–0013 (2008).
Follow the Topic
-
Nature Chemistry
A monthly journal dedicated to publishing high-quality papers that describe the most significant and cutting-edge research in all areas of chemistry, reflecting the traditional core subjects of analytical, inorganic, organic and physical chemistry.


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in