The story behind AMAISE (A Machine learning Approach to Index-free Sequence Enrichment)
Published in Bioengineering & Biotechnology
In 2017, Robert Dickson and other collaborators published work on rapidly identifying bacterial pneumonia using real-time DNA sequencing technology and a metagenomic classification method. Despite this breakthrough, bedside clinical diagnostics based on DNA sequencing technologies and metagenomic classification methods remain uncommon. This is due in part to the fact that current real-time sequencing technologies output long-read data, which is often noisy. Applied to these noisy data, existing metagenomic classification methods are memory intensive and inaccurate.
In 2019, my thesis advisor Jenna Wiens and I started working with Robert Dickson, John Erb-Downward and Piyush Ranjan to determine how machine learning (ML) could improve the accuracy and memory efficiency of the metagenomic classification pipeline. Recognizing that ML methods can perform classification without relying on large reference databases, we first looked into replacing existing metagenomic classification methods with ML methods. Past work has developed ML methods to classify both long read and short read input data. However, outperforming existing non-ML based approaches (e.g., Kraken2, Centrifuge, Minimap2, and Bowtie2) in terms of memory efficiency and accuracy on the task of multi-class classification proved challenging.
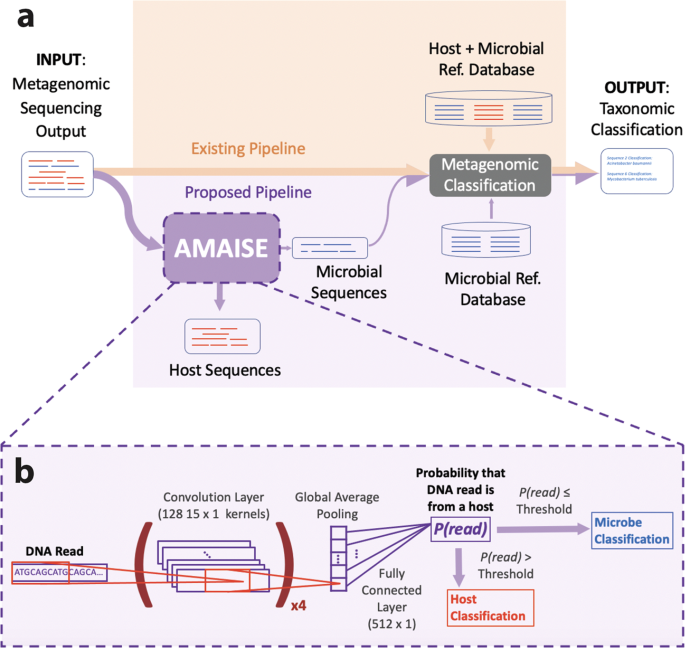
Ultimately, our exploration led us to a realization: much of the inefficiency of existing metagenomic pipelines is due to the necessity to classify (i.e., remove) a large amount of host DNA because many microbiomes are dominated by host data. Thus, instead of entirely replacing existing metagenomic classification methods, we focused on the gains that could be achieved by computationally eliminating host sequences from samples before using them as input to metagenomic classification methods. To this end, we developed, AMAISE (A Machine learning Approach to Index-free Sequence Enrichment) an ML based pre-processing method that computationally removes host data and in turn improves the accuracy and memory efficiency of the metagenomic classification pipeline.
Existing metagenomic classification methods function by identifying exact matches between k-mers within sequences and their reference databases. In contrast, ML methods learn more general patterns. This flexibility provides an advantage when applied to noisier (i.e., long-read) data. Thus, we focus on long-read classification. Given a long-read sequence, AMAISE outputs a classification label determining whether it belongs to a host or a microbe (0 for microbe and 1 for host). When used to augment existing metagenomics pipelines, AMAISE improved accuracy and memory efficiency by over 10% and 14% respectively, while achieving comparable if not better speed to the original pipelines. Furthermore, AMAISE was developed such that it could be applied before time-consuming quality control steps that are typically applied in the current metagenomic classification pipeline.
Accuracy, speed, and memory efficiency are all important in developing bedside clinical diagnostic technologies. High accuracy is critical to ensuring that the correct pathogens are identified and the appropriate treatments selected. Speed ensures that treatment can be given in a timely manner. And memory efficiency is important to the accessibility of the technology. Given the improvements provided by adding AMAISE to the current metagenomic classification pipeline, we believe that this work brings us one step closer to clinical diagnostics via DNA sequencing technologies at the bedside.
Follow the Topic
-
Communications Biology
An open access journal from Nature Portfolio publishing high-quality research, reviews and commentary in all areas of the biological sciences, representing significant advances and bringing new biological insight to a specialized area of research.

Related Collections
With Collections, you can get published faster and increase your visibility.
Artificial Intelligence Methodology in Structural Biology
Publishing Model: Hybrid
Deadline: Nov 30, 2026
Healthy Aging
Publishing Model: Open Access
Deadline: Dec 31, 2026



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in