Triple phase boundary in solid oxide fuel cells: Reaction dynamics based on atomic structure observations
Published in Chemistry
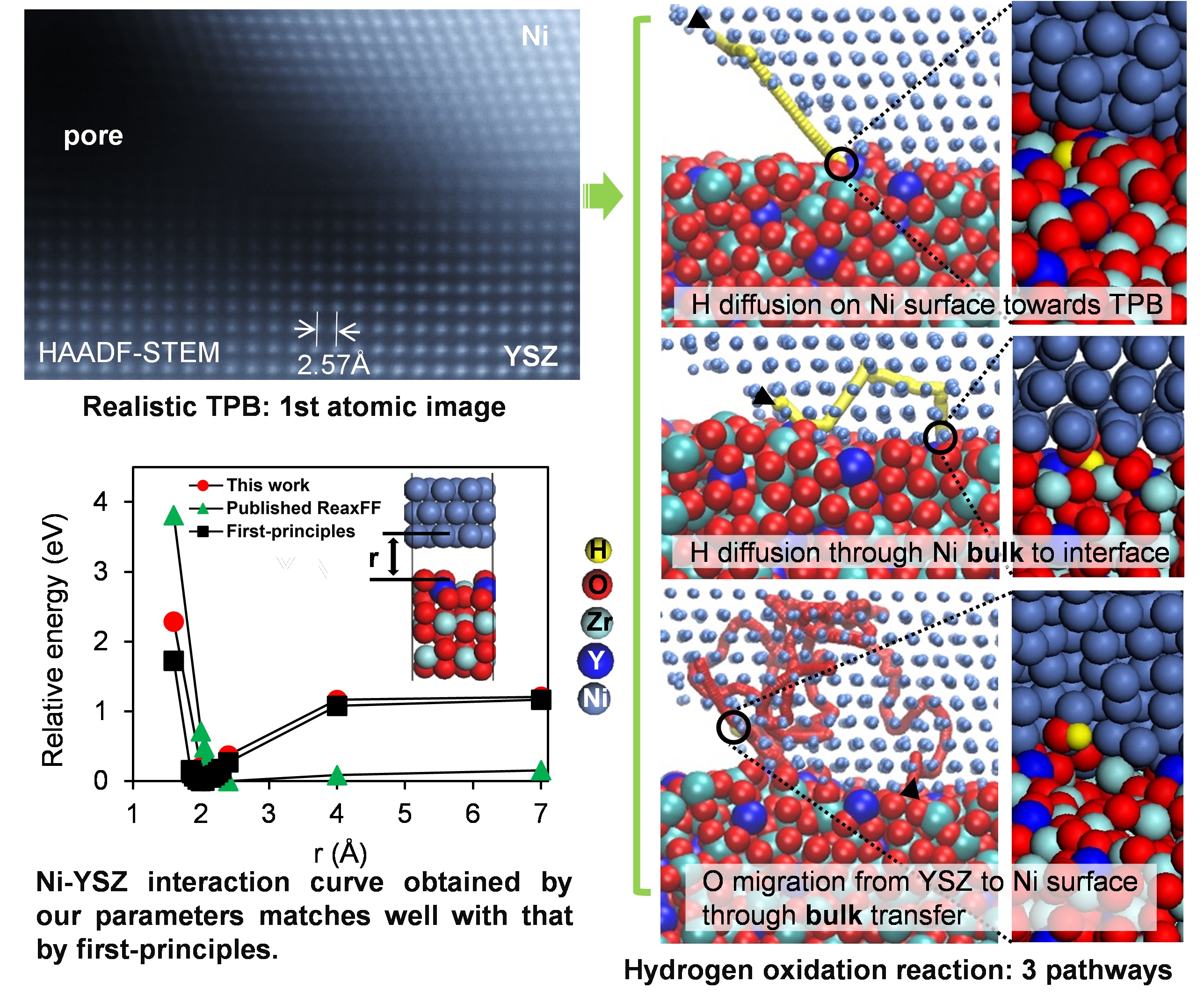

The triple phase boundary (TPB), namely the junction of metal, support, and gas phases, is an extremely important location that affects the material properties, in particularly the catalytic activity of metal/support catalysts, which exist in many forms when they are placed into use, typically nanopowder and bulk. A representative example of the bulk catalysts can be found in the anodes of solid oxide fuel cell (SOFC), where, doubtlessly, Ni/yttria-stabilized zirconia (Ni/YSZ) stands for the standard material. The Ni/YSZ/pore TPB is believed to be the main place for the electrochemical oxidation reaction of fuels.
To estimate the stability of the catalyst and reveal the chemical reaction process at TPB, the atomic structure of TPB is certainly a key for either experimental verification or theoretical simulation. Much attention has been given to nanopowder catalysts on the atomic structures of TPBs in the aspects of both structure observations by using transmission electron microscope (TEM) and theoretical simulations such as ab initio methods, first-principles calculations, and molecular dynamics (MD) simulations. As for the Ni/YSZ bulk anode, which was invented around 50 years ago, however, there has been no report on the realistic atomic structure of TPB. Many efforts have been devoted on TPB, such as the experimental three-dimensional reconstruction on the porous anodes and kinetic modelling on the patterned anodes. However, the spatial scale of those works is limited to submicrometer. The atomic scale works can only be found in the theoretical simulations based on non-realistic models mainly limited by high computational cost for computation. Furthermore, the simulation results from different TPB structure models are often discrepant, which indicates the TPB structure determines the TPB reaction. This study is motivated to explore the reaction pathways at the real Ni/YSZ/pore TPB down to the atomic scale. Herein, we summarize three of our major findings.
First, the atomic resolution observation of the Ni/YSZ/pore TPB to the best of our knowledge is challenged and realized for the first time. We demonstrate our methodology of TEM observation and two TPB structures obtained in this study, which have never been adopted in the previous theoretical studies exploring the reaction mechanism.
Second, large-scale reaction dynamics was challenged. The high computational cost of first-principles methods typically used to study chemical reactions hinders their application to TPB structures observed by atomic resolution microscopy. One possibility is to adopt the reactive force field (ReaxFF), however, previous parameterization appeared to be inaccurate and did not work effectively for the observed TPB structures. Therefore, many efforts have been made to develop a tool to efficiently support the parameterization of ReaxFF and to determine more accurate set of parameters for TPB.
Third, non-conventional HOR mechanisms at TPB are clarified by ReaxFF MD simulations. Distinct reaction pathways and the activity of different structures are identified, which provides a new insight into the understanding of the TPB reactions and verifies the importance of local structure. We would like to emphasize the necessity of the two bulk pathways (H diffusion through Ni bulk to Ni/YSZ interface and O migration from YSZ to Ni surface through bulk transfer) for future researches.

To learn more details, please read our article entitled “Atomic structure observations and reaction dynamics simulations on triple phase boundaries in solid-oxide fuel cells” in Communications Chemistry.
This post was co-authored by Shu-Sheng Liu, Leton C. Saha and Michihisa Koyama.
fuel cell, electron microscope, hydrogen storage, catalysis, thermodynamics
Follow the Topic
-
Communications Chemistry
An open access journal from Nature Portfolio publishing high-quality research, reviews and commentary in all areas of the chemical sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Chemical modification of proteins
Publishing Model: Open Access
Deadline: Sep 30, 2026
Sustainable waste management through polymer upcycling
Publishing Model: Open Access
Deadline: Aug 31, 2026




Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in