Unlocking the Molecular Mechanisms of BSEP Dysfunction: Toward Precision Therapies for PFIC2
Published in Chemistry and Cell & Molecular Biology
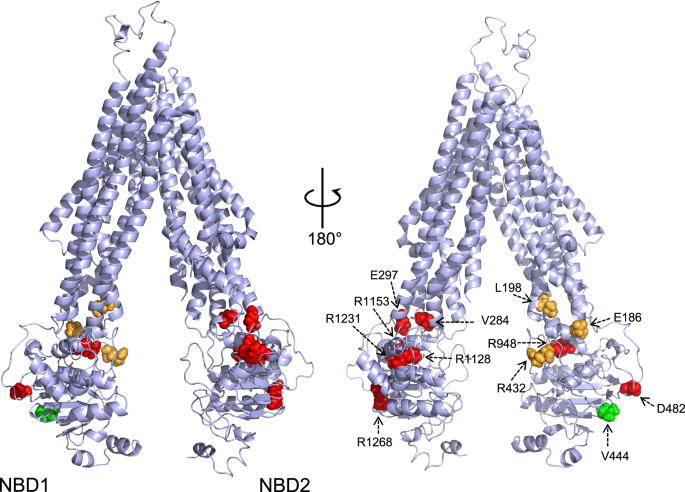
Progressive familial intrahepatic cholestasis type 2 (PFIC2) is a rare but devastating pediatric liver disorder caused by mutations in the bile salt export pump (BSEP, also called ABCB11). Children living with this disorder typically experience intense itching, yellowing of their skin and malnutrition, eventually progressing to end-stage liver disease and often requiring a liver transplant. Like other protein trafficking diseases — including cystic fibrosis, caused by mutations in the ABC transporter CFTR — PFIC2 stems from the failure of a critical membrane protein to reach its correct cellular location and function properly. In the case of PFIC2, the mutated BSEP transporter is dysfunctional, resulting in bile acid retention in hepatocytes and cellular damage.
In protein folding diseases such as these, folding of local structural elements, protein domains, or defective assembly of multidomain proteins may all be affected by missense mutations. Depending on the cellular environment and specific folding pathways, the mechanisms through which misfolded proteins are recognized by the cellular protein quality control machinery and targeted for degradation can vary. Many studies have demonstrated that protein folding defects often lead to a decrease in the thermodynamic stability of the nascently or fully folded protein, and may be observed directly through the experimental determination of the melting temperature of the target protein.
Our work focused on understanding how specific missense mutations in BSEP lead to protein destabilization and trafficking failure, with the hope of identifying druggable hotspots in the protein structure through which trafficking and function might be restored.
Unpacking BSEP Dysfunction at the Molecular Level
Despite growing awareness of protein trafficking disorders in hepatobiliary diseases, including those linked to BSEP and ABCB4, another critical membrane transporter protein implicated in bile acid biology, the molecular details underlying BSEP dysfunction in PFIC2 remained poorly understood.
To address this, we used CETSA (cellular thermal shift assays), immunoblotting, and immunofluorescence to systematically assess a panel of clinically observed BSEP variants. CETSA, a method developed over just the last decade, is capable of determining a protein’s melting temperature within the context of its cellular environment. It has the advantage of characterizing the energetics of folding within the full proteostasis network, including interactions with chaperones or accessory proteins that may be required for proper folding, cellular localization, and function.
These orthogonal approaches revealed a convergent pattern of instability, particularly in a cluster of mutations that localize to the NBD2 region of the BSEP protein— clues that led us to focus on this hotspot of structural instability.
A New Structural Perspective
To gain deeper mechanistic insight, we determined the cryo-EM structure of a pathogenic mutant BSEP (E297G) at high resolution. The structure revealed disorder in the NBD2 region, supporting the biochemical data and offering a potential explanation for impaired trafficking and function. This is, to our knowledge, the first cryo-EM structure of a clinically relevant BSEP mutant, and it provides a valuable framework for understanding how mutations perturb ABC transporter folding.
Looking Ahead
Through biophysical, immunofluorescence, and structural analyses, we have demonstrated a clear correlation between the thermodynamic stability of BSEP variants, their cellular localization, and the severity of associated diseases. Notably, our CETSA experiments have identified significant destabilization at the NBD2-ICL2 interface, a critical region for protein folding and function. Furthermore, the cryo-EM structure of the BSEP E297G variant provides direct structural confirmation of significant disorder in NBD2, likely as a result of this highly destabilizing mutation.

The discovery of hotspots for destabilization within ABC transporters supports the notion that certain protein classes are, by nature, poised at the edge of stabilization. These proteins require low energy barriers for transitions between conformational states to function effectively, which, for ABC transporters such as BSEP, involves interconversion between inward and outward-facing conformations to facilitate the transport of substrates across biological membranes. While necessary for function, this inherent instability may also make these regions susceptible to destabilizing mutations that can lead to disease.
By mapping specific destabilizing mutations and visualizing their structural consequences, our work offers a path toward precision therapeutics that could restore BSEP trafficking and function for patients with PFIC2 and potentially for additional hepatobiliary diseases characterized by dysfunction in bile salt transport, such as primary sclerosing cholangitis.
These findings have the potential to inform the rational design of small molecule correctors and potentiators that can restore and enhance transporter function. More broadly, they contribute to efforts to address diseases caused by membrane protein misfolding at the molecular level.
Follow the Topic
-
Communications Biology
An open access journal from Nature Portfolio publishing high-quality research, reviews and commentary in all areas of the biological sciences, representing significant advances and bringing new biological insight to a specialized area of research.

Related Collections
With Collections, you can get published faster and increase your visibility.
Artificial Intelligence Methodology in Structural Biology
Publishing Model: Hybrid
Deadline: Nov 30, 2026
Healthy Aging
Publishing Model: Open Access
Deadline: Dec 31, 2026

Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in