Unveiling genome evolution of the invasive common reed
Published in Ecology & Evolution, Protocols & Methods, and Genetics & Genomics
The common reed is a grass family species that seems to thrive just about anywhere across the globe. Known for its remarkable resilience, this species appears to flourish not only in dry, barren lands but also in harsh, salty and alkalic soils. European lineage of common reed was introduced to North America around 200 years ago, since then and especially within the past 50 years it has become notorious for its invasiveness. Interestingly, the original source population in Europe hasn’t exhibited the same invasive behavior. This puzzling change in life style has led researchers to explore the factors underlying its rapid transformation into an invasive species. Dspite this interest the underlying genomic changes driving this shift have remained largely unexplored due to the absence of high-quality genome assemblies.

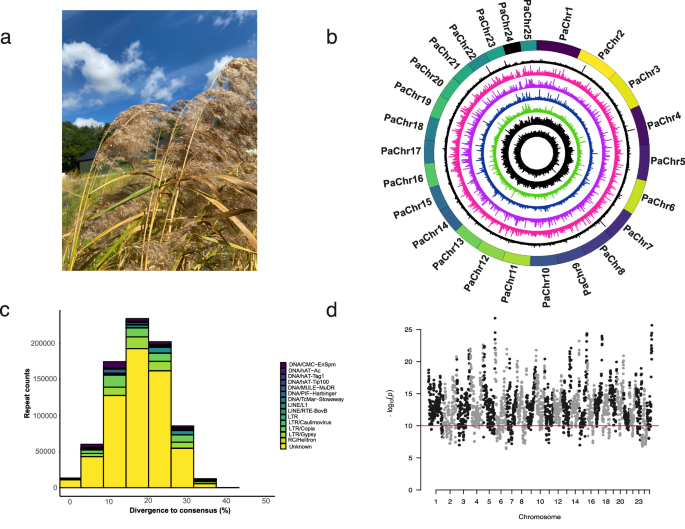
The rapid progress of genome sequencing techniques has revolutionized our ability to study genomes in detail. Our team began gathering data for Pacbio HiFi long read sequencing in 2021, and combining that data with chromosome capture information (Hi-C) ultimately yielded a gap-free, telomere-to-telomere (T2T) chromosome-level genome assembly for the common reed. Although the common reed is allotetraploid, that is, a hybrid of two distinct plant species, we were thrilled to be able to assign all the pseudochromosome assemblies into either one of the two subgenomes. We first presented the assembly at an SMBE Regional Meeting on the Role of the Genome in Biological Invasion in February 2023.
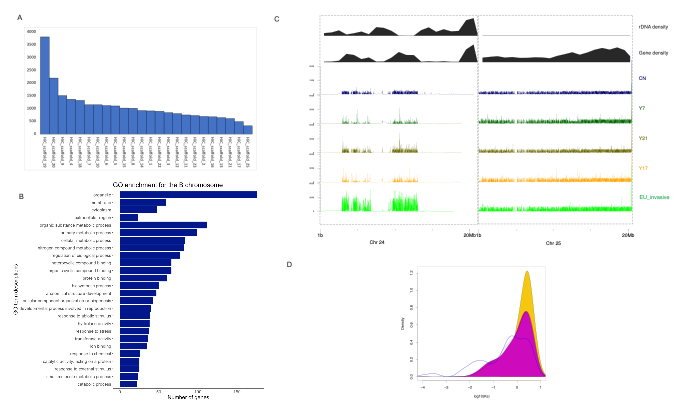
Our excitement was further increased when we discovered an unexpected extra chromosome in the assembly. The common reed was expected to have 24 haploid chromosomes, but we assembled 25. We first tested whether this extra chromosome truly existed within the cells. We prepared karyotype slides and, to our astonishment, observed 50 chromosomes. Upon analyzing the genome content of this additional chromosome we found it to be mostly composed of repetitive regions and fragments inserted from other 24 chromosomes. We therefore concluded that we had sequenced the B chromosome of common reed. This discovery was particularly exciting as gap-free T2T sequences for B chromosomes are rare—only a few species have had their B chromosomes sequenced, with maize being one of the most recent ones in 2021. The discovery of a B chromosome in the common reed opens new avenues for understanding its genome evolution. By analyzing whole genome sequencing read coverages in five individuals representing other lineages, we found that the invasive representative had higher coverage on the B chromosome and may thus possess more copies of this chromosome compared to other representatives. Intriguingly, our analysis revealed that the genes predicted on B chromosome were enriched for with gene ontology terms related to gene expression regulation, RNA synthesis, and telomere maintenance. In addition, we identified a high number of tandem duplications in the invasive lineage, suggesting their potential role in the development of invasiveness.
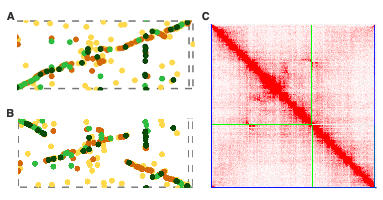
Our journey with the common reed genome had also other challenges. For instance, when doing the final assembly, we noticed that chromosome 22 displayed a different synteny when aligned to the rice genome compared to our previous version. Curiously, the Hi-C figure appeared perfect in both instances. This eventually led us to find out that Chr22 harbors a haplotypic inversion. Observing such dynamic genome structures within the common reed genome is fascinating, and we speculate that its clonal nature may contribute to maintaining these unique feature.
As we continue to delve into the genome of this resilient and invasive grass, each discovery brings about new questions and an ever-growing sense of wonder at the complexity of nature.
Follow the Topic
-
Communications Biology
An open access journal from Nature Portfolio publishing high-quality research, reviews and commentary in all areas of the biological sciences, representing significant advances and bringing new biological insight to a specialized area of research.

Related Collections
With Collections, you can get published faster and increase your visibility.
Artificial Intelligence Methodology in Structural Biology
Publishing Model: Hybrid
Deadline: Nov 30, 2026
Healthy Aging
Publishing Model: Open Access
Deadline: Dec 31, 2026



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in