Unveiling the Mysteries of the CTCF R567W Mutation in Neurodevelopmental Disorders
Published in Cell & Molecular Biology and Genetics & Genomics
Motivation and Research Journey
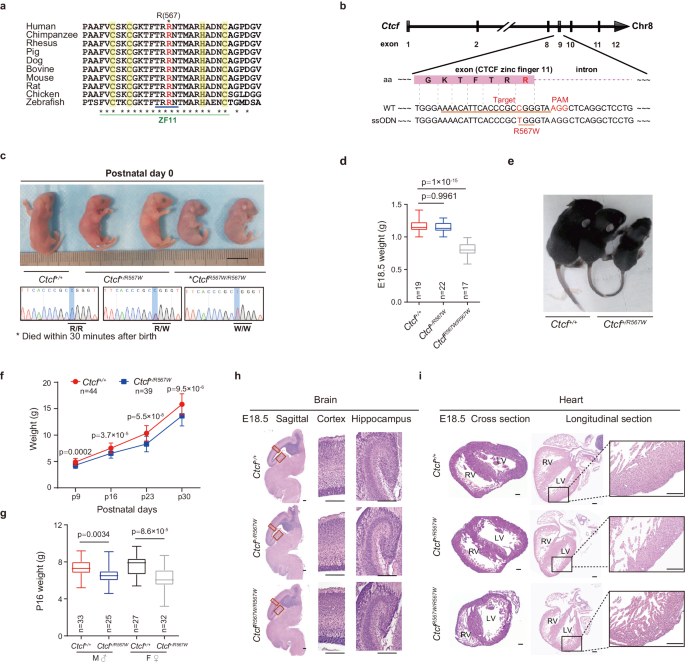
Our journey began with a puzzling clinical observation - children harboring a specific mutation in the master genomic architect, CTCF. This single alteration led to the transformation of the arginine at position 567 to a tryptophan (R567W). These children exhibited various neurodevelopmental abnormalities, ranging from intellectual disabilities and growth deficiencies to autistic behaviors and congenital heart defects. This disruption in the complex three-dimensional folding of DNA orchestrated by CTCF, which controls gene expression, seemed to throw the entire developmental program into disarray. This single misstep in the genetic dance exerted profound consequences.
Driven by curiosity and a desire to unravel the underlying mechanisms, our team began an extensive quest, developing models that recapitulated this impactful mutation. Given the limited clinical samples available for direct examination of such mutations, we introduced the R567W mutation into mouse genome, observing ripple effects from the earliest stages of development. Simultaneously, we harnessed the capability of human embryonic stem cells (hESCs), coaxing them to form cortical organoids that served as a window into the human condition.
Key Findings
Initially, we were frustrated that, although we tried extensive behavioral experiments, the results showed no obviously phenotypic abnormalities in heterozygous CTCF R567W mutant mice. This disappointed us as our mouse model did not fully recapitulate the neurodevelopmental abnormalities observed in clinical cases. However, in the homozygous state, the consequences were severe – mice succumbed to early postnatal mortality, and their development irreversibly derailed. At embryonic day 18.5 (E18.5), these embryos displayed notable neurological and cardiopulmonary development abnormalities, foreshadowing this single mutation's profound impact.
Within the complex structure of the cerebral cortex, at the single-cell level, the CTCF-R567W mutation unleashed a cascade of cellular events. Stem-like cells were prematurely depleted, and the maturation of GABAergic neurons was accelerated, disrupting the equilibrium required for correct neurodevelopment. Synaptic pathways, the intricate networks enabling neuronal communication, were thrown into disarray, preceding the cognitive and behavioral abnormalities that would later be observed. Consistent with the mouse model, the hESC-derived cortical organoid model demonstrated that the homozygous CTCF-R567W mutation hindered self-organization during differentiation. In contrast, at the single-cell level, even the heterozygous mutation was sufficient to produce an imbalance in the differentiation of stem-like cells and neuron maturation, reflecting the clinically abnormal phenotype.
However, the true power of our investigation was observed in the molecular mechanisms at play. The R567W mutation, a seemingly innocuous change, disrupted CTCF's ability to bind to certain genomic regions, particularly those harboring the upstream motifs. This disruption in binding caused a reorganization of chromatin interactions, reshaping the three-dimensional landscape of the genome and changing gene expression patterns. One locus emerged as a pivotal player – the clustered protocadherin (cPcdh) genes, essential for neuronal survival and synaptic connectivity. The CTCF-R567W mutation orchestrated a profound remodeling of chromatin architecture, modulating long-range interactions and isolating specific regions into distinct topologically associating domains (TADs). The consequences of this reorganization were clear in the transcriptional landscape, as the expression of cPcdh genes was significantly dysregulated, contributing to the observed neurodevelopmental defects.
Our findings surpassed the boundaries of model systems, as the human cortical organoids mirrored the cellular and molecular perturbations identified in the mouse models. This convergence of evidence highlighted the profound impact of the CTCF-R567W mutation on neurodevelopment, a testament to the conserved and essential role of CTCF in governing the genomic landscape across species.

Significance and Future Outlook
With all of the success observed, the journey has only just begun. The alignment of chromatin conformational changes at the individual neuron level with patterns identified in bulk cells merits further exploration. Additionally, highlighting the tissue-specific pathologies and cross-talk between genetic and environmental factors will be essential in revealing the full spectrum of CTCF-related disorders.
Moreover, our study represents a pivotal step toward developing innovative therapeutic strategies. Utilizing human-derived organoid models, we can screen for compounds that restore the delicate balance of chromatin folding, paving the way for precision medicine approaches for individual CTCF mutations.
As we move toward this exciting future, one truth becomes evident: the genome is a masterpiece of design, and CTCF, the maestro orchestrating its activity, holds the key to unlocking the mysteries of human development and disease. Our study elucidates the crucial role of CTCF in neurodevelopmental disorders and paves the way for further investigations into the complexities encoded in our genetic blueprint.
Follow the Topic
-
Nature Communications
An open access, multidisciplinary journal dedicated to publishing high-quality research in all areas of the biological, health, physical, chemical and Earth sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Women's Health
Publishing Model: Hybrid
Deadline: Ongoing
Tumor Microenvironment Crosstalk and Therapeutic Implications
Publishing Model: Hybrid
Deadline: Nov 02, 2026


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in