What happens in the Eastern Pacific stays in the Eastern Pacific – the genomic basis of adaptive evolution in three-spined sticklebacks revisited
Published in Ecology & Evolution


In this study we show that the patterns of genetic parallelism in marine-freshwater ecotype differentiation in three-spined sticklebacks (Gasterosteus aculeatus) is unique and exceptional in the Eastern Pacific compared to other parts of the world. Since three-spined sticklebacks in the Eastern Pacific are an almost iconic example of “parallel evolution” in the wild, it is important to understand that what is happening in the Eastern Pacific is unusual, perhaps even exceptional compared to other species and model systems. This possibility has intrigued me for a long time, ever since I used an example data set from three-spined sticklebacks to demonstrate the merits of a novel unsupervised analysis method to detect loci involved in parallelism and local adaptation back in 2015.

Linkage disequilibrium network analyses
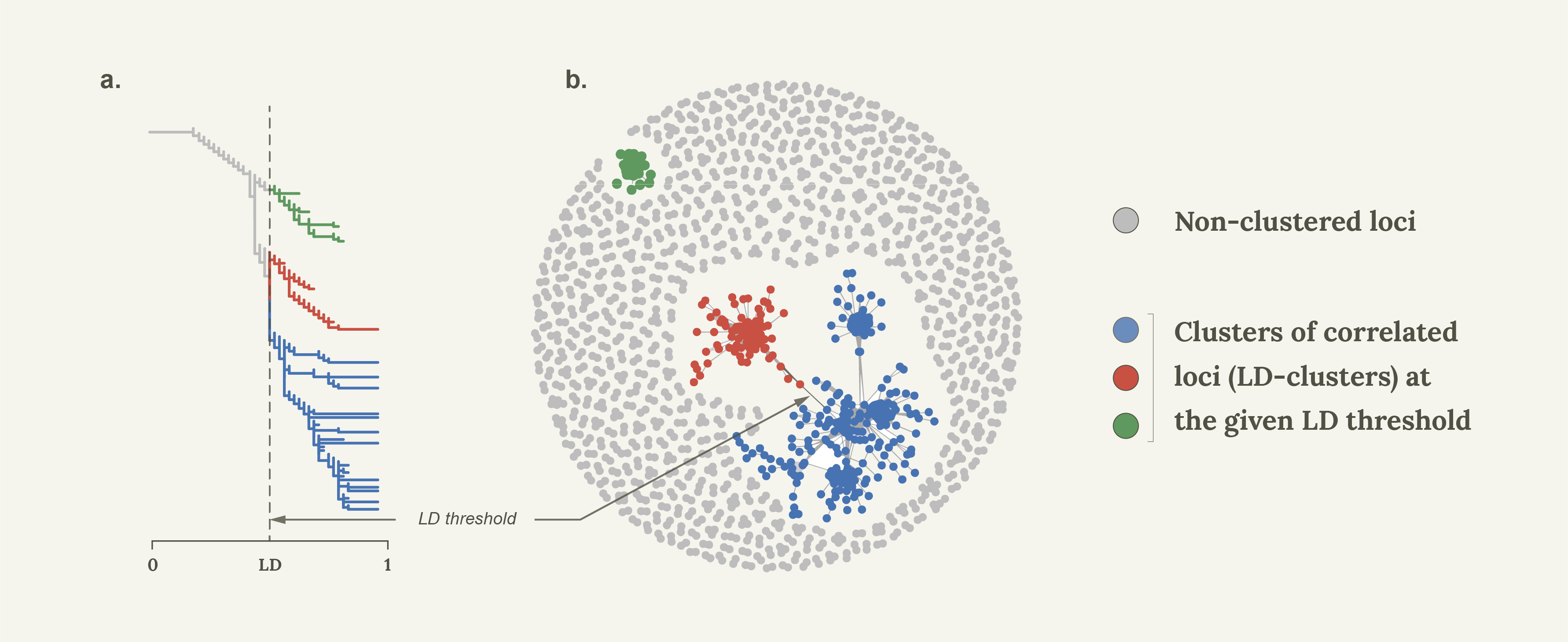
Let’s start with some basics. Linkage disequilibrium (LD) is the non-random association of alleles between pairs of loci. It’s common estimate, r2, ranges between 0 (linkage equilibrium) and 1 which basically means that one locus can completely replace another without changing the data set. It turns out that much like clustering/network analytical methods are used to assess phylogenetic relationships between individuals, so can they be used to infer correlation patterns between loci based on LD.
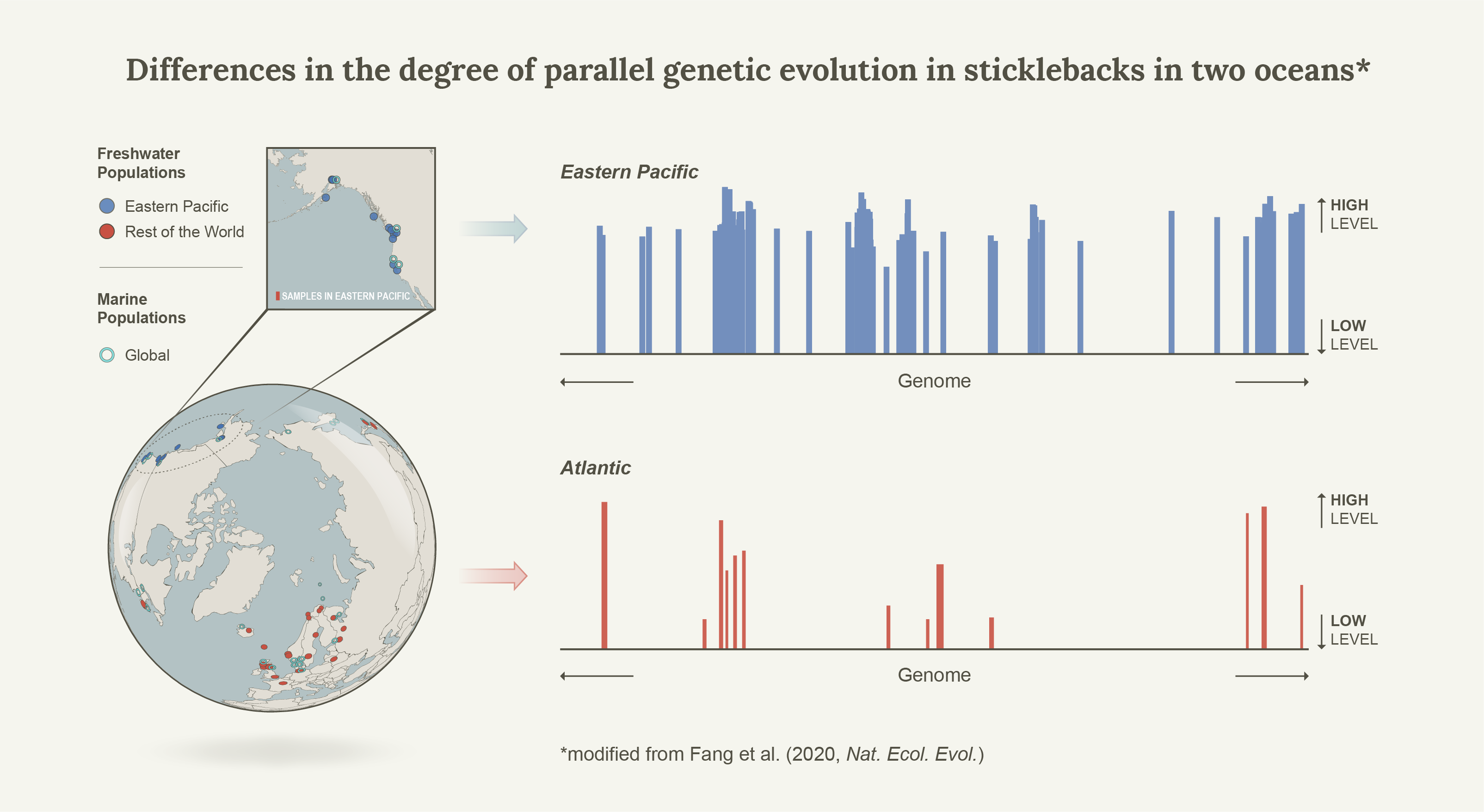
In 2015, together with colleagues I showed that Linkage Disequilibrium network analyses (LDna) can be used to separate population genomic data into sets of highly correlated loci that reflect distinct evolutionary phenomena such as population demographic history, chromosomal rearrangement (most notably inversions) and local adaptation1. As proof of concept we used a subset of loci from 21 genomes from Jones et al.’s2 seminal paper on the genomics of local adaptation in three-spined sticklebacks. Indeed, in a single analysis we detected distinct LD-clusters that were associated with three known inversions and geographic separation between Pacific and Atlantic samples, but also found a surprisingly large set of loci that separated all freshwater individuals from the Eastern Pacific from all other individuals (that is, Pacific marine individuals as well as marine and freshwater individuals from the Atlantic). Thus, rather than clustering all freshwater individuals from all marine individuals (global parallelism) these loci seemed to show ecotype differentiation exclusively in the Eastern Pacific (local parallelism). We concluded that much of the global patterns of marine-freshwater divergence could be driven by what was happening in the Eastern Pacific, but this was never followed up until now. It turns out that this was only the beginning of the story.
Sticklebacks as a model system to study evolution
By a coincidence I ended at the university of Helsinki and eventually joined Juha Merilä’s research group in 2018. Our main interest is understanding the role of demographic history on local adaptation and genetic parallelism. While the three-spined stickleback is the most famous model system among the numerous other species in the Gasterosteus genus – and indeed one of the most important model systems to study evolutionary biology in the wild overall – the much less studied nine-spined stickleback (Pungitius pungitius) has considerably stronger population structuring and is often found landlocked in very small and isolated post-glacial ponds across the northern hemisphere. Since local adaptation (and thus also genetic parallelism) heavily depends the access to standing genetic variation, we expect much less of this in nine- compared to three-spined sticklebacks. Collectively these two species comprise an important model system for comparative evolutionary biology.
As we sought to compare the levels of genetic parallelism between three- and nine-spined sticklecakcs, we initially focused on the Atlantic region, where we had access to good geographic sampling for both species. With the general consensus at the time – that genetic parallelism is exceptional in the three-spined sticklebacks – we expected to find large clusters of loci associated with marine-freshwater differentiation in the three-spined sticklebacks (but not in the nine-spined sticklebacks). To my initial surprise we did not, which had me doubt the data set and our methodology. Consequently, we took a step back and included a number of Eastern Pacific samples as well, including the ones used in the example data set in Kemppainen et al.1, but a much larger set of SNPs. Indeed, the cluster that separated all freshwater individuals from the Eastern Pacific from all other individuals was still there. How could this be?
Back to the drawing board
Well, while most studies of marine-freshwater genetic parallelism were conducted on Eastern Pacific populations (demonstrating genome wide and strong genetic parallelism), the few that were starting to emerge from the Atlantic showed a different pattern. While indeed all genomic regions showing marine-freshwater differentiation comprised a subset of those found also in the Easter Pacific (consistent with the colonization history of this species), the regions were much fewer and covered much smaller regions of the genome. Furthermore, we eventually realized that this pattern was also present in Jones et al.2, but since the focus was on global parallelism this pattern was never mentioned in the paper. They used a self-organizing map-based iterative Hidden Markov Model (SOM/HMM) that, similarly to LDna, sorted the genome into sets that represent a given evolutionary tree, and one of the largest (comprising 2.83% of the genome) indeed separated Eastern Pacific freshwater individuals from all the rest. Thus, the discrepancy was not only apparent in our analyses, but was consistent with all of the previous literature as well. The focus of the comparative study quickly turned back the three-spined sticklebacks, and to fully understand what really was happening.
What happens in the Eastern Pacific stays in the Eastern Pacific
Since the Atlantic Basin was colonised during the last opening of the Bering strait (~40 k years ago3,4) and freshwater adapted alleles are generally only found in low frequencies in the sea, it was natural to consider a scenario where a substantial proportion of the Pacific ancestral variation never reached the Atlantic due to founder events. This was tested with simulations and indeed, consistent with the empirical data (where the expected reduction in neutral genetic variation in the Atlantic also was seen), the proportion of the genome that was affected by parallel marine-freshwater differentiation was a function of trans-oceanic gene flow during the colonization of the Atlantic Basin (from the Eastern Pacific). It was also a function of QTL density; while high QTL density in low recombination regions was a pre-requisite for “differentiation islands” to form, the combined selection against of such tightly linked QTL also meant that the density of the freshwater alleles for these QTL were the lowest in the sea (and thus were also the most affected by founder events). This was surprising but in hindsight makes sense.
Secondary contact in the Eastern pacific?
The simulation results were nevertheless not entirely consistent with the empirical data and could for instance not explain why the marine Eastern Pacific individuals in the empirical data were genetically more similar to the marine and freshwater individuals from the Atlantic than to freshwater individuals from their own region. Long geographic isolation (i.e. random fixation of alternative alleles due to drift) is the most common source of LD in population genomic data sets, pointing to the fact that some form of geographic isolation followed by secondary contact might also have played a role in creating such a strong discrepancy in the patterns of marine-freshwater differentiation between Eastern Pacific and the rests of the world. This hypothesis, presented for the first time by Bierne et al.5, has received surprisingly little attention in the three-spined stickleback literature. We know that large “ice-lakes” that were not connected to the sea have existed on the Northern American continent throughout much of it’s geological history, dating back to the time when three- and nine-spined sticklebacks diverge from each other some 26 million years ago. Thus, if there was a long period of geographic isolation between marine and freshwater populations, this would have resulted in not only adaptive differences but also neutral. If the marine and freshwater populations in the Eastern Pacific became connected to each other only after the Atlantic basin was colonised, the large majority of ecotype differences (adaptive as well as neutral) in the Eastern Pacific would remain unique to this area. While we have not yet tested this hypothesis by simulations (we did not want to prolong the publication of this study more than necessary), we do think that there is a lot of merit to this hypothesis, and I’m looking forward to seeing how this research field may potentially spark some new fire in light of our recent findings.
To sum up – the paper did not emerge overnight from nothing. The bare bones of the story – or the discovery – was hatched years ago, but a few coincidences allowed me return to dig deeper into this with the enthusiastic support of our team with most of the hard work of analysing and putting together the final manuscript being done by Bohao Fang (first author).
A note on methodology
The SOM/HMM method of Jones et al.2. differs from LDna on a few important accounts. First, it requires full genome sequence data which we did not have, since most of our data set comprised of high-density SNP data. Second, in a study by Li et al.6. we showed that the genome cannot be considered as a continuous stretch of DNA that support one phylogenetic pattern or the other, but instead comprises of sets of correlated SNPs that can be interspersed and overlap along chromosomes. Since LDna considers each locus pair individually (in no particular order) it can potentially lead to a much higher resolution of the different evolutionary phenomena that have shaped the genetic variation in the genome. The major drawback of LDna is of course that it considers all pairwise LD values between loci in the data set at once, so cannot be used for very large data sets. This was solved in Li et al.6. by firstly finding correlated SNPs along non-overlapping windows along the chromosomes, and only using one of the SNPs from each cluster for the next face of the analyses, resulting in highly efficient complexity reduction with minimal loss of information. Linkage Disequilibrium network analyses were then conducted on each chromosome at a time. Then, using one SNP from each of these clusters, a final LDna was conducted pulling together correlated LD-clusters from different chromosomes. Indeed, despite that we did not analyse full genomes, we recovered a large proportion of all the regions that were involved in global parallelism in Jones et al.2, thus demonstrating that most of the information in the data could be summarised by the few “tag” SNPs that represented each cluster from the first LDna step.
References
- Kemppainen, P. et al. Linkage disequilibrium network analysis (LDna) gives a global view of chromosomal inversions, local adaptation and geographic structure. Mol. Ecol. Resour. 15, 1031-1045, doi:10.1111/1755-0998.12369 (2015).
- Jones, F. C. et al. The genomic basis of adaptive evolution in threespine sticklebacks. Nature 484, 55-61, doi:10.1038/nature10944 (2012).
- Fang, B. et al. Estimating uncertainty in divergence times among three-spined stickleback clades using the multispecies coalescent. Molecular Phylogenetics and Evolution 142, 106646 (2020).
- Fang, B. et al. Worldwide phylogeny of three-spined sticklebacks. Molecular Phylogenetics and Evolution127, 613-625 (2018).
- Bierne, N. et al. The geography of introgression in a patchy environment and the thorn in the side of ecological speciation. Current Zoology 59, 72-86 (2013).
- Li, Z. et al. Linkage disequilibrium clustering-based approach for association mapping with tightly linked genomewide data. Mol. Ecol. Resour. 18, 809-824, doi:10.1111/1755-0998.12893 (2018).
I would like to thank shared first author Bohao Fang (@fangbohao) and co-authors Paolo Momigliano (@PaoloMomigliano) and Juha Merilä for the opportunity to pursue this project. This was truly a team effort with everyone contributing crucially to shape the final product through several major revisions and re-analyses.
Follow the Topic
-
Nature Ecology & Evolution
This journal is interested in the full spectrum of ecological and evolutionary biology, encompassing approaches at the molecular, organismal, population, community and ecosystem levels, as well as relevant parts of the social sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Understanding species redistributions under global climate change
Publishing Model: Hybrid
Deadline: Dec 31, 2026




Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in