Mechanochemistry as a Tool of Polymer Physics
Published in Chemistry

Dilute solutions of polymers have countless uses in everyday life, laboratory research and industry. Unlike small-molecule solutes, dissolved macromolecules are very sensitive to how and how fast the solution flows, responding to such flows in complex ways, from adopting new 3D shapes at low flow rates to undergoing chemical reactions that permanently change their properties, including breaking chains, in fast flows.1 The latter is called flow-induced mechanochemistry and is too often a detrimental process, responsible for considerable costs and economic losses. Successful attempts to suppress, or even exploit, flow-induced mechanochemistry remain rare, stymied by our lack of detailed understanding of how solvent flows alter macromolecular properties.2
In a major breakthrough in the late 90s Steve Chu and colleagues at Stanford developed a means of observing individual superlong DNA molecules in gently flowing solutions.3 These observations challenged a number of then-prevalent ideas of how a flowing solution affects its macromolecular solutes and launched the field of molecular rheology. Yet, as much as this approach shaped our thinking about polymer solutes, it has proven impossible to extend to flows of practical or conceptual interest. As a result, by now we understand far better the behavior of fairly unusual polymers in esoteric flows than of conventional polymers in common flows.
Our recently appeared Nature Chemistry paper “Experimental quantitation of molecular conditions responsible for flow-induced polymer mechanochemistry”4, reports a complementary approach that works best at the very fast rather than gentle solvent flows. These are produced by cavitation and hitherto were thought to be least amenable to detailed characterization. Cavitation occurs in mind-bogglingly diverse scenarios, capable of destroying both ship propellers and kidney stones (in lithotripsy) and can be generated by a variety of mechanisms, including by sonication, i.e., by passing sound waves of certain frequency used in our work. Cavitation involves the formation and violent collapse of microscopic gas bubbles. At each implosion, a thin layer of liquid around the bubble gets accelerated to very high flow rates, albeit for only a microsecond or so. Such flow rates are enough to stretch dissolved macromolecules beyond their breaking points. Because the underlying processes are very fast, and at any moment only a tiny fraction of the dissolved macromolecules are close enough to the collapsing bubbles to experience rapidly flowing solvent, the conventional optically tracking of individual macromolecules in these violent but transient flows is impossible.
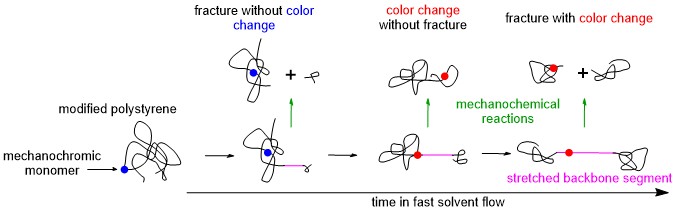
Instead, we quantified their dynamics by measuring mechanochemistry of polystyrenes containing a single “mechanochromic” monomer per backbone (Figure above). When stretched hard enough these polystyrenes break, just as any other macromolecule does, but they can also change color without breaking or both break and change color. The probability of each outcome depends on how the stretching force experienced by each monomer changes as the polymer backbone interacts with the flow. That’s because each chain could break at many sites along its backbone, but change its color only when its sole mechanochromic monomer experienced a sufficient stretching force.
We prepared batches of modified polystyrenes of different lengths, with the mechanochromic monomer located at precisely known locations that varied very little within each sample but ranged from a chain terminus to the chain center among the samples. This allowed us to quantify, with the help of mathematical modelling, how the geometries of reacting chains evolved in the rapidly accelerating solvent flows around them by simply measuring the fractions of chains that broke with and without changing color, and changed color without breaking (see animation).
This approach extends experimental molecular rheology to practically-interesting flows and common macromolecules, and reduces the resolution at which chain geometries are quantifiable down to 10 nm and sub-microseconds. It shows that conditions that are completely incompatible with optical spectroscopy, and are therefore invisible to the established methods, are still amenable to detailed molecular characterization. At present, extracting the distributions of molecular conditions responsible for mechanochemical reactions of individual chains from measured changes in bulk compositions of cavitating polymer solutions remains resource-intensive, which may limit the adoption of our method. We are hard at work streamlining this data processing. In the meantime, the potential of our approach to upend our understanding of chain behavior in solvent flows is already obvious, as illustrated by three surprising conclusions that emerged from our proof-of-the-approach studies.
- One of the most unexpected findings of Chu et al was how resistant dissolved macromolecules were to stretching from a nearly-spherical blob of constantly fluctuating dimensions (a “random coil”) to a straight rod. Indeed, the latter has rarely, if ever, been observed experimentally, the fact usually attributed to the experimentally observable flows being too slow and/or too transient. Such complexities were thought to be irrelevant in the much faster flows that cause mechanochemistry, i.e., by the time a chain becomes mechanochemically reactive, it was believed to have been stretched to a rod-like shape. Our measurements refute this comforting simplicity, suggesting that only 20 – 30% of the backbone of fragmenting chains is appreciably stretched, although we don’t know how close to a rod-like geometry the rest of the backbone is. In these flows chains appear to be orders of magnitude slower to equilibrate internally than the existing models of molecular relaxation dynamics require. As a result, the force experienced by one monomer of such a chain has little effect on the forces experienced by most other monomers, which allows a very inert C-C bond to break without causing much more labile monomers elsewhere in the chain to react as well.
- Bulk measurements (unlike single-molecule alternatives) usually reflect ensemble-averaging, which obscures the variation in conditions experienced by individual molecules. Our work demonstrates that important information about the distributions of molecular conditions are still inferable from bulk measurements: we quantified the distributions of molecular geometries of fractured chains, the stretching force they experienced at the moment of fracture and the time they spent stretched before fracturing. We succeeded because a macromolecule in a flow can break at many different locations along the backbone, and the relative contribution of each monomer is highly sensitive to the local conditions of the flow. A sonicated solution contains tens of thousands of unique macromolecules, with their own combinations of chain length, location of the mechanochromic monomer and its color, and we could track the evolution of each such component. As a result, we identified subpopulations of chains each resulting from unique reaction conditions and from the relative abundance of these subpopulations we derived the probability of each set of molecular conditions to occur. While these aspects may seem unique to mechanochemistry in sonicated solutions, we suggest that opportunities to extract distributions of conditions experienced by individual reacting molecules from cleverly designed bulk measurements may be more general than is currently thought.
- Our work both exploited polymer mechanochemistry and allowed us to test experimentally some of the ideas that currently inform the design of mechanochemical experiments and mechanoresponsive molecules.5 Perhaps most consequential for the design of practical mechanoresponsive polymers is our finding that placing mechanosensitive monomers (“mechanophores”) away from the chain center increases both the selectivity of productive mechanochemistry and its rate. Our discovery that force acting on a reacting monomer changes as fast as the monomer reacts suggests that the loading rate (how fast force changes) may be a more promising parameter to exploit in designing hitherto unknown patterns of mechanochemical reactivity than the magnitude of the force.
- Larson, R. G., The rheology of dilute solutions of flexible polymers: Progress and problems. J. Rheology 2005, 49, 1-70.
- O'Neill, R. T.; Boulatov, R., The many flavours of mechanochemistry and its plausible conceptual underpinnings. Nature Rev. Chem. 2021, 5, 148-167.
- Perkins, T. T.; Smith, D. E.; Chu, S., Single polymer dynamics in an elongational flow. Science 1997, 276, 2016-2021.
- O'Neill, R. T.; Boulatov, R., Experimental quantitation of molecular conditions responsible for flow-induced polymer mechanochemistry. Nature Chem. 2023.
- Akbulatov, S.; Boulatov, R., Experimental polymer mechanochemistry and its interpretational frameworks. Chemphyschem 2017, 18, 1422-1450.
Follow the Topic
-
Nature Chemistry
A monthly journal dedicated to publishing high-quality papers that describe the most significant and cutting-edge research in all areas of chemistry, reflecting the traditional core subjects of analytical, inorganic, organic and physical chemistry.



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in