
The discovery of p53, famously dubbed the ‘guardian of the genome’, has revolutionized our understanding of how normal and cancer cells grow and divide as well as the protein’s importance for cancer protection [1]. More than twenty years of research has shown that p53 is a sequence-specific transcriptional activator that responds to genotoxic stimuli (e.g. DNA damage, oncogene activation, oxidative stress or hypoxia) and protects the genome by inducing a variety of biological responses including DNA repair, cell cycle arrest, senescence, apoptosis or autophagy [2]. Moreover, it plays a role in several non-canonical cellular processes, such as modulating autophagy and regulating metabolism [3]. Importantly, we now know that over half of human cancers harbor missense mutations in p53 that inhibit its function, making it the most mutated protein in cancer [4]. This knowledge has since paved the way for understanding the role of other related genes and pathways in cancer initiation and progression, as well as identifying key targets in the development of cancer therapeutics [3].
Rather unexpectedly, studies have reported that a subset of p53 mutants self-assemble into inactive cytosolic amyloid-like aggregates [5, 6], suggesting that the associated cancers can be classed as amyloid diseases. Amyloid diseases are disorders that are associated with misfolding and self-assembly of functional proteins or peptides into amyloids, which are aggregates that are characterized by a fibrillar morphology, a predominantly β-strand secondary structure, high thermodynamic stability, insolubility in common solvents and resistance to proteolytic digestion [7-9]. These amyloid diseases include Alzheimer’s disease, type II diabetes, Parkinson’s disease, transmissible spongiform encephalopathies (or prion diseases) and Huntington’s disease [8, 9].
p53 is a homotetramer with each monomer composed of a globular, thermodynamically unstable DNA-binding domain (DBD), which houses the vast majority (~90%) of cancer-associated mutations, separated by a flexible linker to a tetramerization domain [3, 10]. These mutations hamper the protein’s transcriptional regulation role by either inhibiting p53’s capacity to interact with target DNA (contact mutants), or by disrupting the protein’s three-dimensional structure (structural mutants) [11]. However, various so-called p53 ‘contact’ mutations, including the commonly occurring R248W and the closely related R248Q, also cause alterations in the structure and conformational stability of the protein [12, 13]. Destabilizing the protein’s structure, in turn, results in exposure of the DBD’s hydrophobic core and self-assembly of the protein into amyloid-like aggregates within inactive cellular inclusions [14, 15]. These inclusions often include wild-type (WT) p53 as well as its paralogs (p63 and p73), leading to a loss of WT p53’s tumor suppressor function and suppression of the regulatory functions of p63 and p73 [16].
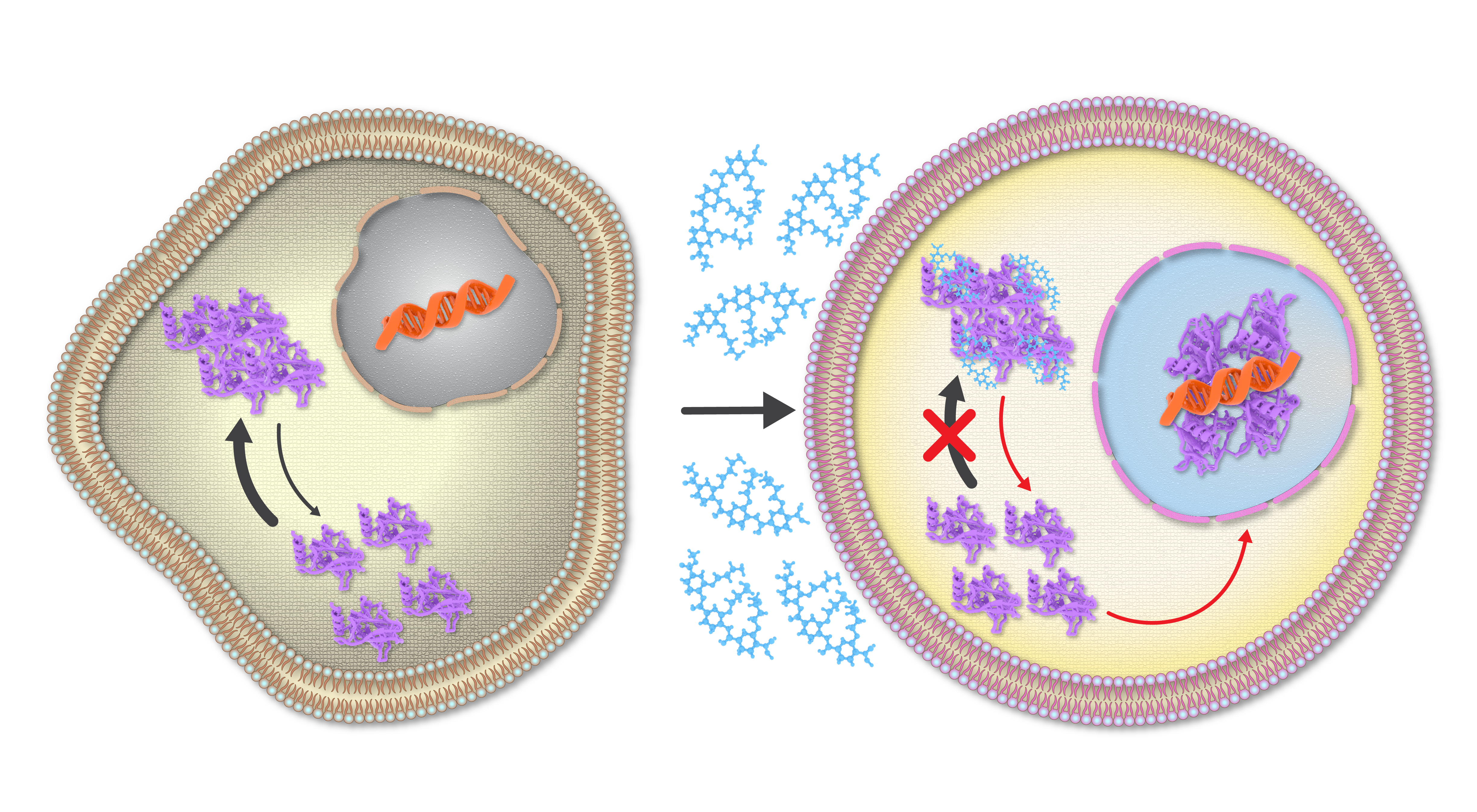
Surprisingly, only a few studies to-date have investigated therapeutic strategies targeting mutant p53 amyloid-like aggregation. The most notable of these is the sequence-specific peptide inhibitor, ReACp53, that has been shown to block mutant p53 aggregation by masking the DBD’s aggregation-nucleating subdomain [17]. Given our lab’s interest in both amyloid diseases and cancer [18, 19], we decided to leverage our expertise to identify and/or develop potent mutant p53-targeted cancer therapeutics. We began by screening a focused library of protein α-helix mimetics, previously shown to antagonize amyloid assembly of the amyloid-β peptide (Aβ) and islet amyloid polypeptide (IAPP), which are associated with Alzheimer’s disease and type II diabetes, respectively [19, 20]. From this screen, we identified a protein mimetic, ADH-6, that potently inhibits oligomerization and amyloid formation of a mutant p53 DBD-derived peptide containing both the aggregation-nucleating sequence and the commonly occurring R248W mutation [21]. ADH-6 also dissociates pre-formed aggregates of the peptide and prevents its further aggregation, suggesting that the protein mimetic can disaggregate intracellular mutant p53 amyloid-like aggregates. This was confirmed by studies in human cancer cells harboring mutant p53, which showed that ADH-6 targets and dissociates intracellular mutant p53 amyloid-like aggregates more effectively than ReACp53 [21].
But what makes the protein mimetic a more potent amyloid inhibitor than ReACp53? NMR studies showed that ADH-6 interacts with not only the aggregation-nucleating subdomain, but also several other regions of mutant p53 DBD, whereas ReACp53 only binds to the aggregation-nucleating subdomain of the DBD [21]. This enables ADH-6 to effectively target and stabilize both the monomeric and pre-amyloid α- helical states of mutant p53 and prevent its amyloid aggregation, which strongly shifts the folding equilibrium of the protein towards the soluble state, resulting in dissociation of the protein’s inactive amyloid-like cytosolic aggregates [21].
Subsequent studies, done in collaboration with colleagues at NYU Abu Dhabi, NYU New York and the University of Denver, showed that ADH-6 mediated dissociation of mutant p53 amyloid-like aggregates in human cancer cells restores p53’s transcriptional activity, leading to cell cycle arrest and apoptosis [21]. ADH-6 also displayed substantially longer in vivo circulation half-life and prolonged presence in the bloodstream compared to ReACp53. This can be attributed to the fact that synthetic protein mimetics, with their constrained backbone, possess greater inherent in vivo stability than peptides [22, 23]. We speculated that the extended in vivo circulation time of ADH-6 would facilitate increased accumulation in tumor tissue. Indeed, this was confirmed by our studies, which showed that ADH-6 treatment more effectively reduces the size of xenografts harboring mutant p53 compared to ReACp53, while exhibiting no toxicity to healthy tissue, thereby substantially prolonging survival [21].  Figure 1. The protein mimetic amyloid inhibitor ADH-6 efficiently enters cancer cells to directly interact with and dissociate mutant p53 amyloid-like aggregates, leading to restoration of p53’s transcriptional activity.
Figure 1. The protein mimetic amyloid inhibitor ADH-6 efficiently enters cancer cells to directly interact with and dissociate mutant p53 amyloid-like aggregates, leading to restoration of p53’s transcriptional activity.
By focusing on a largely ignored property of many p53 mutants, namely their propensity to aggregate into inactive clusters, we have demonstrated the first successful application of a bona fide small-molecule amyloid inhibitor as an anticancer agent. We therefore believe this work will have a broad impact as it effectively establishes a bridge between amyloid diseases and cancer, providing a foundation for cross-informational approaches in the design of new and potent mutant p53-targeted cancer therapeutics.
Written by Mona Kalmouni, Palanikumar Loganathan & Mazin Magzoub.
References
-
- Efeyan, A. and M. Serrano, p53: guardian of the genome and policeman of the oncogenes. 2007. 6(9): p. 1006-1010.
- Teodoro, J.G., S.K. Evans, and M.R. Green, Inhibition of tumor angiogenesis by p53: a new role for the guardian of the genome. 2007. 85(11): p. 1175-1186.
- Kastenhuber, E.R. and S.W. Lowe, Putting p53 in context. 2017. 170(6): p. 1062-1078.
- Hollstein, M., et al., p53 mutations in human cancers. 1991. 253(5015): p. 49-53.
- Silva, J.L., et al., Prion-like aggregation of mutant p53 in cancer. 2014. 39(6): p. 260-267.
- Silva, J.L., et al., Targeting the prion-like aggregation of mutant p53 to combat cancer. 2018. 51(1): p. 181-190.
- Sunde, M. and C.C. Blake, From the globular to the fibrous state: protein structure and structural conversion in amyloid formation. 1998. 31(1): p. 1-39.
- Caughey, B. and P.T. Lansbury Jr, Protofibrils, pores, fibrils, and neurodegeneration: separating the responsible protein aggregates from the innocent bystanders. 2003. 26(1): p. 267-298.
- Chiti, F. and C.M. Dobson, Protein misfolding, functional amyloid, and human disease. 2006. 75: p. 333-366.
- Krois, A.S., H.J. Dyson, and P.E. Wright, Long-range regulation of p53 DNA binding by its intrinsically disordered N-terminal transactivation domain. 2018. 115(48): p. E11302-E11310.
- Demma, M., et al., SCH529074, a small molecule activator of mutant p53, which binds p53 DNA binding domain (DBD), restores growth-suppressive function to mutant p53 and interrupts HDM2-mediated ubiquitination of wild type p53. 2010. 285(14): p. 10198-10212.
- Muller, P.A. and K.H. Vousden, Mutant p53 in cancer: new functions and therapeutic opportunities. 2014. 25(3): p. 304-317.
- Joerger, A.C. and A.R. Fersht, The p53 pathway: origins, inactivation in cancer, and emerging therapeutic approaches. 2016. 85: p. 375-404.
- Xu, J., et al., Gain of function of mutant p53 by coaggregation with multiple tumor suppressors. 2011. 7(5): p. 285-295.
- Soragni, A., et al., A designed inhibitor of p53 aggregation rescues p53 tumor suppression in ovarian carcinomas. 2016. 29(1): p. 90-103.
- Bom, A.P.A., et al., Mutant p53 aggregates into prion-like amyloid oligomers and fibrils: implications for cancer. 2012. 287(33): p. 28152-28162.
- Zhang, Y., et al., Therapeutic potential of ReACp53 targeting mutant p53 protein in CRPC. 2020. 23(1): p. 160-171.
- Palanikumar, L., et al., pH-responsive high stability polymeric nanoparticles for targeted delivery of anticancer therapeutics. 2020. 3(1): p. 1-17.
- Henning-Knechtel, A., et al., Designed cell-penetrating peptide inhibitors of amyloid-beta aggregation and cytotoxicity. 2020. 1(2): p. 100014.
- Maity, D., et al., Sub-stoichiometric inhibition of IAPP aggregation: a peptidomimetic approach to anti-amyloid agents. 2020. 1(4): p. 225-232.
- Palanikumar, L., et al., Protein mimetic amyloid inhibitor potently abrogates cancer-associated mutant p53 aggregation and restores tumor suppressor function. Nature Communications, 2021. 12(1): p. 3962.
- Azzarito, V., et al., Inhibition of α-helix-mediated protein–protein interactions using designed molecules. 2013. 5(3): p. 161-173.
- Jayatunga, M.K., et al., α-Helix mimetics: Outwards and upwards. 2014. 24(3): p. 717-724.
Follow the Topic
-
Nature Communications
An open access, multidisciplinary journal dedicated to publishing high-quality research in all areas of the biological, health, physical, chemical and Earth sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Women's Health
Publishing Model: Hybrid
Deadline: Ongoing
Biosensing
Publishing Model: Hybrid
Deadline: Sep 30, 2026

Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in