DeepMainmast: Integrated Protocol of Protein Structure Modeling for cryo-EM with Deep Learning and Structure Prediction
Published in Materials, Protocols & Methods, and Computational Sciences
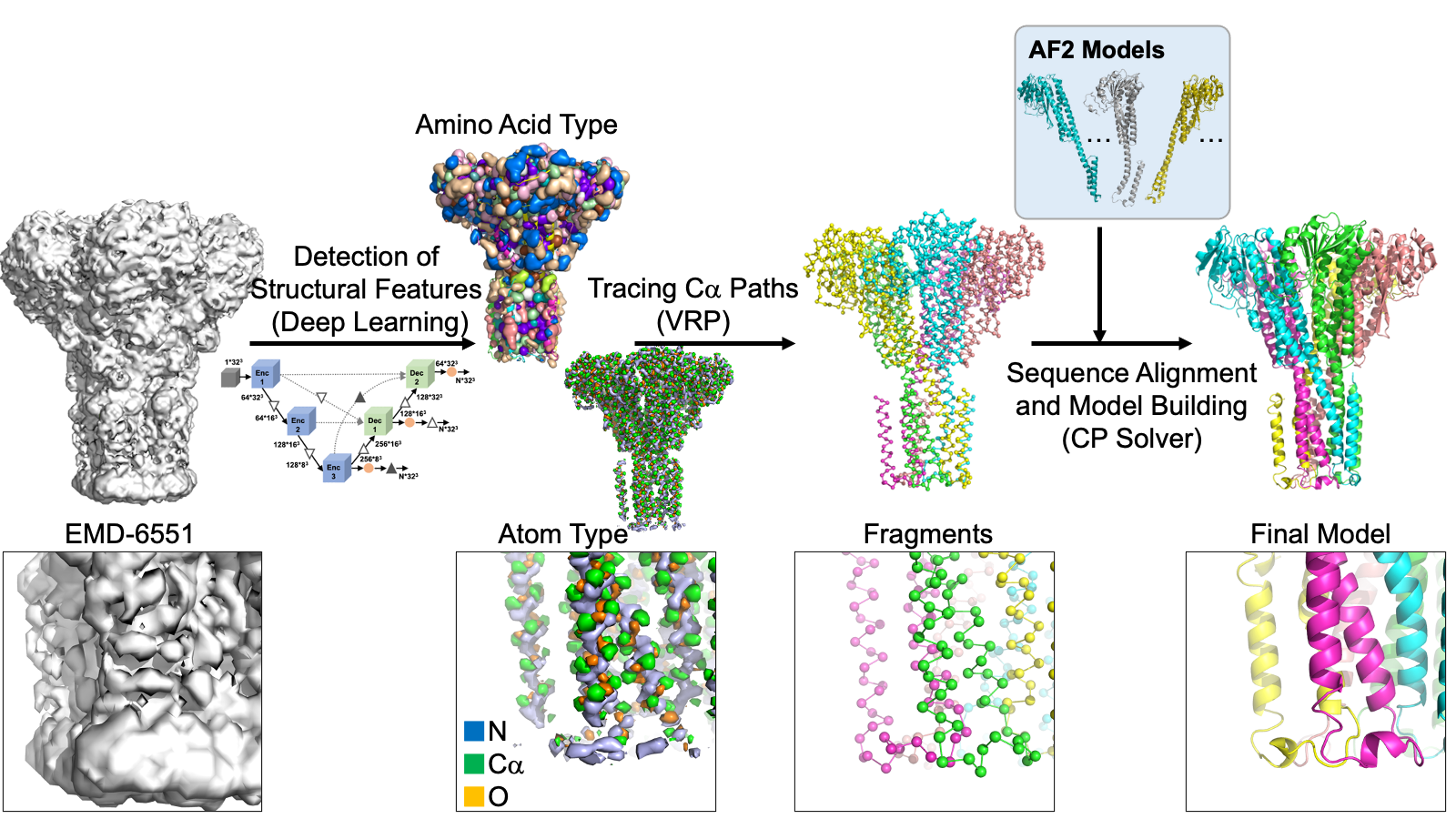
Background
Building 3D models of proteins from cryogenic electron microscopy (cryo-EM) maps is a complex task. Model building is particularly difficult when the map resolution is worse than around 2 to 3 Å when the protein main-chain is not easily traced manually. Resolutions between 3 to 5 Å are particularly frustrating as they reveal parts of the protein structure but lack full clarity. To address this, a team of researchers from Purdue University, Genki Terashi, Xiao Wang, Devashish Prasad, Tsukasa Nakamura, and Prof. Daisuke Kihara, has introduced a groundbreaking method called DeepMainmast. This method merges deep learning to identify key atoms, problem-solving techniques to link these atoms, and Alphafold2 (AF2), a tool for predicting protein structures. This integration significantly enhances the accuracy of protein structure modeling.
DeepMainmast protocol
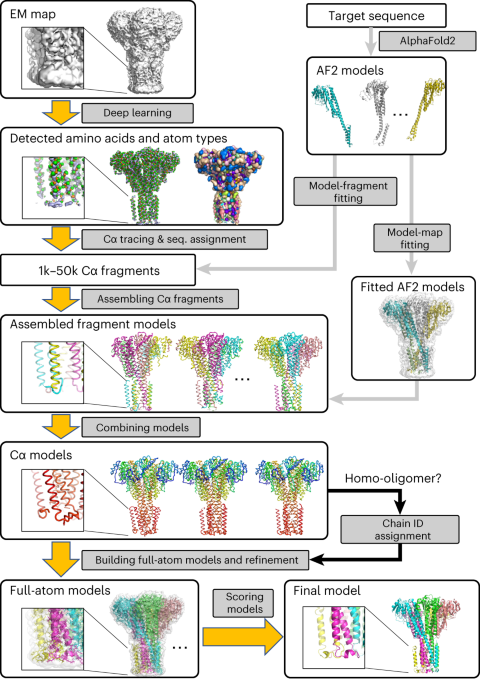
The modeling process begins with using deep learning to spot protein main-chain and side-chain atoms within a cryo-EM map. Then, it utilizes two powerful problem solvers—the Vehicle Routing Problem (VRP) solver and the Constraint Programming (CP) Solver—to link these identified atoms. These solvers connect the atoms while ensuring that the resulting fragments resemble realistic protein chains. Typically, 1000 to 50,000 structural fragments are generated for a protein complex in a map. Next, according to the predicted amino acid type at each of the detected Calpha atoms, protein sequences are assigned to each of the structural fragments. At this step, fragments taken from AF2 are also incorporated into the fragment collection as long as the AF2 fragments have some level of agreement with traced fragments from the cryo-EM map. Subsequently, fragments are combined by the CP solver to build the entire structure of the protein complex. The resulting models are refined and ranked based on their fit to the cryo-EM map and overall quality. DeepMainmast is equipped with a specific procedure for accurate chain identity assignment for homo-multimers, which is not trivial as all homomer chains have identical sequences. Figure 1 illustrates the overall protocol of the DeepMainmast pipeline.

The development of DeepMainmast started a few years ago, and AF2 appeared in the research community during the development. Although the core framework of the DeepMainmast algorithm was developed over a couple of years ago, coming up to the effective strategy to integrate AF2 into the protocol and to achieve significantly improved modeling performance over AF2 took time. During the benchmark study compared to existing methods, weaknesses in the initial version of DeepMainmast were identified. Addressing these weaknesses and fine-tuning the protocol required a substantial amount of time, too. It was great teamwork with Dr. Xiao, an experienced tool developer and the main developer of CryoREAD, which is a DNA/RNA modeling method; Devashish, an energetic and skilled undergraduate student; Dr. Tsukasa, a new postdoc who recently joined our lab, and Professor Kihara, who leads our lab.
Future direction
We have recently developed an RNA/DNA structure modeling protocol called CryoREAD . This fully automated method identifies the positions of phosphate, sugar, and base in a cryo-EM map through deep learning, which are then traced and modeled into a three-dimensional structure using essentially the same protocol as DeepMainmast. A combination of DeepMainmast and CryoREAD for modeling protein–nucleic acid complexes from cryo-EM maps is on the way.
Availability
A DeepMainmast webserver is available at https://em.kiharalab.org/algorithm/DeepMainMast for easy access and use. The full source code is available on GitHub at https://github.com/kiharalab/DeepMainMast. Additionally, users can access the Google Colab Notebook web server at https://colab.research.google.com/github/kiharalab/DeepMainMast/blob/main/DeepMainMast.ipynb. If you have any questions, please contact Prof. Daisuke Kihara at dkihara@purdue.edu.
Follow the Topic
-
Nature Methods
This journal is a forum for the publication of novel methods and significant improvements to tried-and-tested basic research techniques in the life sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Methods development in Cryo-ET and in situ structural determination
Publishing Model: Hybrid
Deadline: Oct 30, 2026



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in