Dissecting the RNA structurome, one conformation at a time
Published in Protocols & Methods
Among all the molecules that build-up a living cell, RNA is definitely (one of) the most fascinating and intriguing. Not only RNA can behave as a carrier of information to drive the synthesis of proteins, but also it can fold back on itself, creating the most amazing and intricate structures. These RNA structures are involved in almost any biological process, both physiological and pathological.
RNA structures are stabilized thanks to the ability of bases within the RNA chain to interact with each other, just like it happens in between the two helices composing DNA. Therefore, within a given RNA structure certain bases will be paired, while some others will be unpaired (or single-stranded).
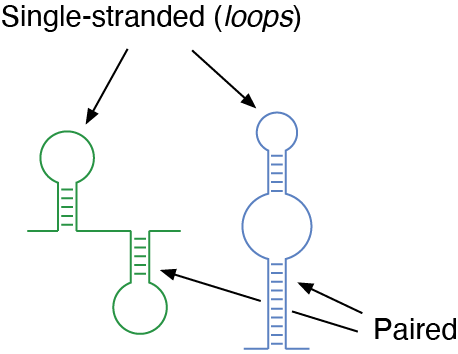
Schematic representation of RNA structures. Certain regions are paired, by direct interaction of complementary bases within the RNA chain, while other regions are single-stranded (commonly referred to as loops).
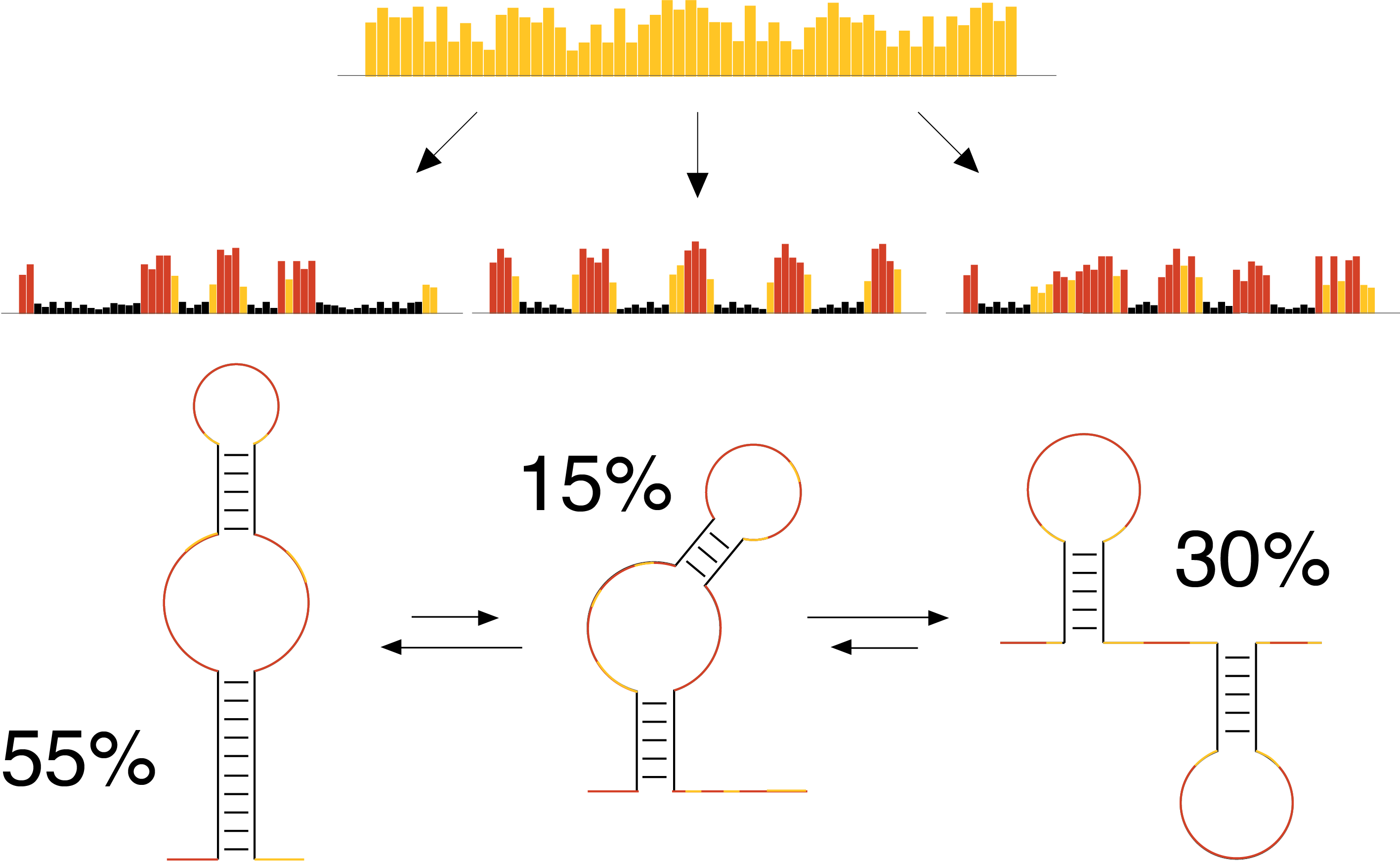
RNA structures are highly diverse and dynamic. Inside the cell, the same RNA is present in multiple identical copies, and not all of them will fold into the same structure. Rather, many alternative structures (or conformations) for the same RNA coexist in a dynamic equilibrium. In addition to that, each conformation is not static over time, but it can interconvert between any other potential conformation for that RNA. This structurally heterogeneous set of RNAs is commonly referred to as an ensemble.
Within the ensemble, each possible conformation for a given RNA is associated with a certain probability of being formed (or observed). This probability is determined (and influenced) by a multitude of factors, including (but not limited to) temperature, concentration of ions and interaction with other molecules. In layman’s terms: if an RNA can form 2 mutually-exclusive conformations, “A” and “B", the first having a probability of 20% and the second having a probability of 80%, at any time, if one would take a picture of an ensemble of 100 RNA molecules, (s)he would see ~20 A-shaped molecules and ~80 B-shaped molecules.
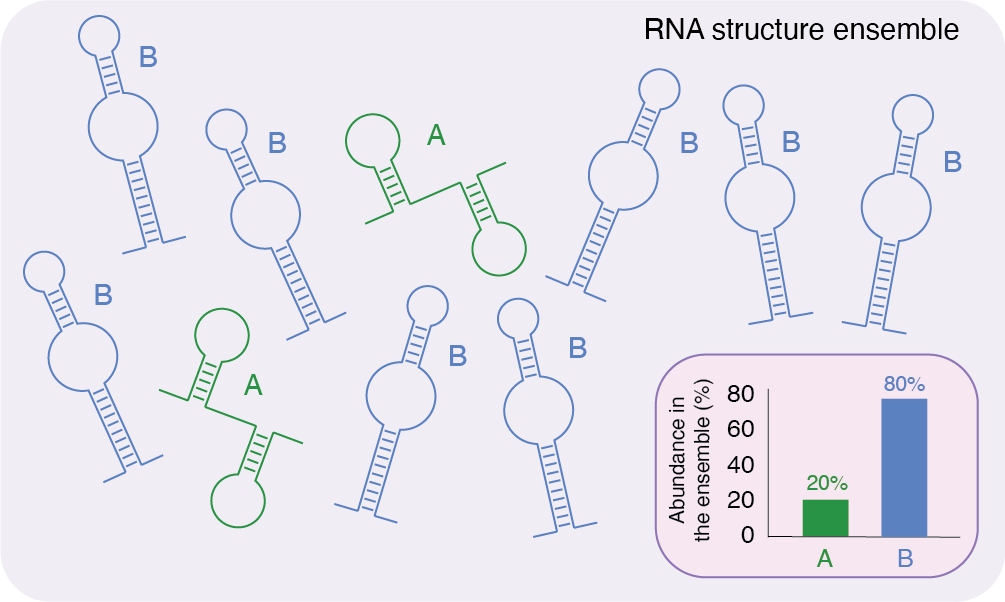
Schematic representation of a hypothetical RNA structure ensemble. Several copies of the same RNA molecule can fold into different conformations. In this example, the RNA has a 20% probability of folding into conformation A, and an 80% probability of folding into conformation B.
At this point one might argue, why is this important? Well, for example, different conformations can mediate different functions. This is for example the case of the 5’-most portion of the HIV-1 RNA genome, whose dynamics control different stages of the viral life cycle. Alternatively, the ability to switch from one conformation to another can control the ability of the cell to use the information contained in the RNA molecule. This is the case of riboswitches, RNA structures that can interconvert between two conformations (ON and OFF) to control the translation of the RNA in which they are contained, by regulating the accessibility of the ribosome to its cognate binding site. These RNA elements represent a crucial mechanism of regulation in bacteria.
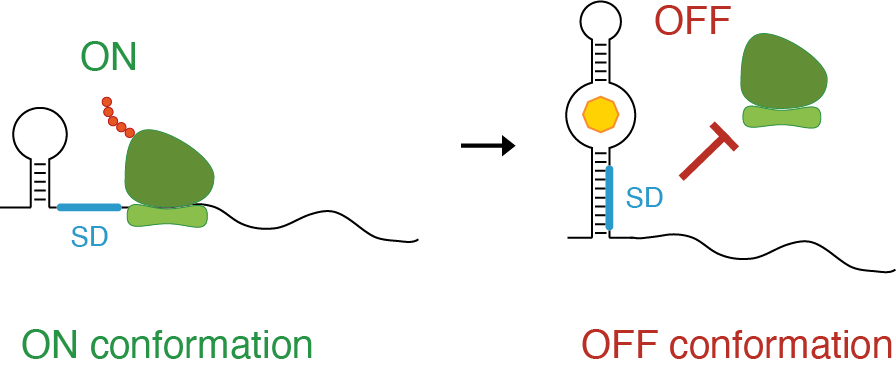
Example of riboswitch. One structural conformation leaves the ribosome binding site (Shine-Dalgarno, SD) accessible (single-stranded), hence allowing translation of the RNA (ON). The other conformation, prevents the ribosome from accessing its binding site, hence impeding translation (OFF).
Ok, so, what is the challenge of “visualizing” such RNA structural dynamics? One of the most commonly employed methods for studying RNA structures is known as RNA probing. In RNA probing experiments, special chemicals are used to modify the single-stranded regions of folded RNA molecules, while paired bases will be protected. Then, by using a reaction called reverse transcription, that enables the conversion of an RNA molecule into a DNA molecule, it is possible to read these sites of chemical modification, hence obtaining a map of the single-stranded bases in the RNA structure. This kind of map can (in)directly inform us on the structure formed by a given RNA. And here comes the real challenge. As we previously discussed how the RNA is heterogeneous and dynamic within the ensemble, it becomes immediately evident that, when we want to interrogate the structure of an RNA using the aforementioned chemicals, we will simultaneously query all the possible conformations that compose the ensemble for that RNA. As a consequence, the map we will get from these experiments would just represent a weighted average of all the coexisting RNA conformations. Just like for Chinese shadow puppetry, what we see is just a shadow generated by the superposition of multiple entities and, as such, it can be highly misleading.
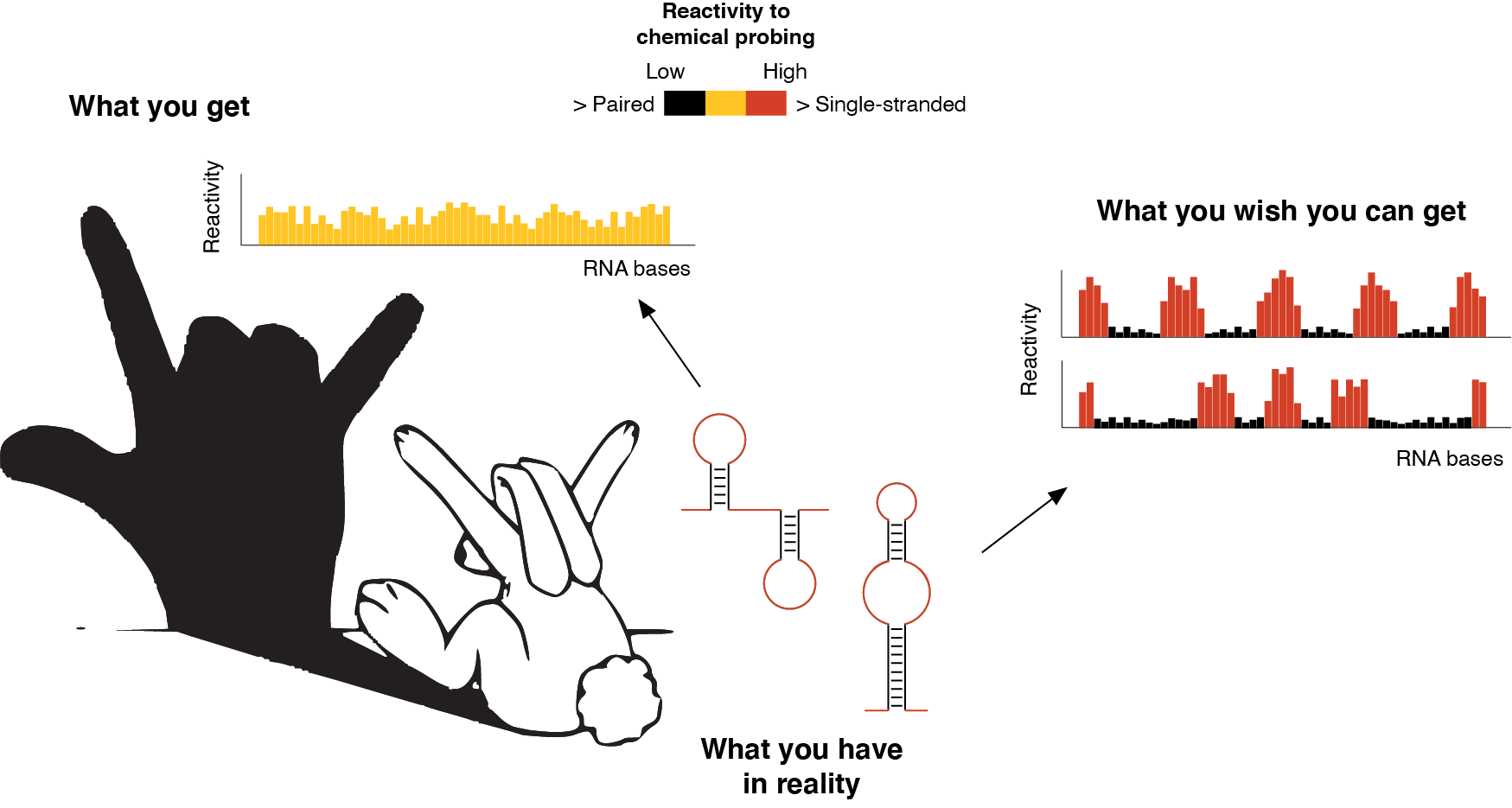
RNA probing is like Chinese shadow puppetry. What we see is just the “projected shadow” of what we really have. In the above example, an RNA can fold into two mutually-exclusive alternative conformations. The signal measured by RNA probing (the shadow) will be the weighted average of the signal coming from both conformations. However, what we actually need to do is to be able to measure the reactivity of each conformation, independently.
Back in 2017, me and my friend (and colleague) Edoardo (Morandi) started discussing about whether it would have been possible to deconvolute the individual structures of the coexisting conformations for a given RNA, by using RNA probing. By then, several groups already tried to approach the problem by using different computational strategies, but the solution was definitely not trivial, and most of the proposed methods lacked the robustness to allow the analysis of large RNA molecules. However, among them, we found that one method in particular, developed by the lab of Prof. Weeks at UNC Chapel Hill, had a significant potential for further improvement. Underlying assumption of the method was that, it is possible during the reverse transcription step to record multiple sites of chemical modification, corresponding to bases that were simultaneously single-stranded within the same original RNA molecule, as mutations within the same resulting DNA molecule, an approach dubbed mutational profiling. As two mutually-exclusive conformations are characterized by different combinations of single-stranded and paired bases, it is theoretically possible to look at the patterns of co-mutation in the reverse-transcribed DNA molecules to determine whether more than one conformation for the studied RNA existed in the ensemble, and the relative abundance of each conformation. For instance, if bases 11 and 18 are single-stranded and base 22 is paired in conformation A, while bases 11 and 22 are single-stranded and base 18 is paired in conformation B, two possible DNA molecules will be generated at reverse transcription: one with bases 11 and 18 mutated, and one with bases 11 and 22 mutated. However, no DNA molecule will have both bases 18 and 22 mutated, indicating that two mutually-exclusive conformations are present in the sample.
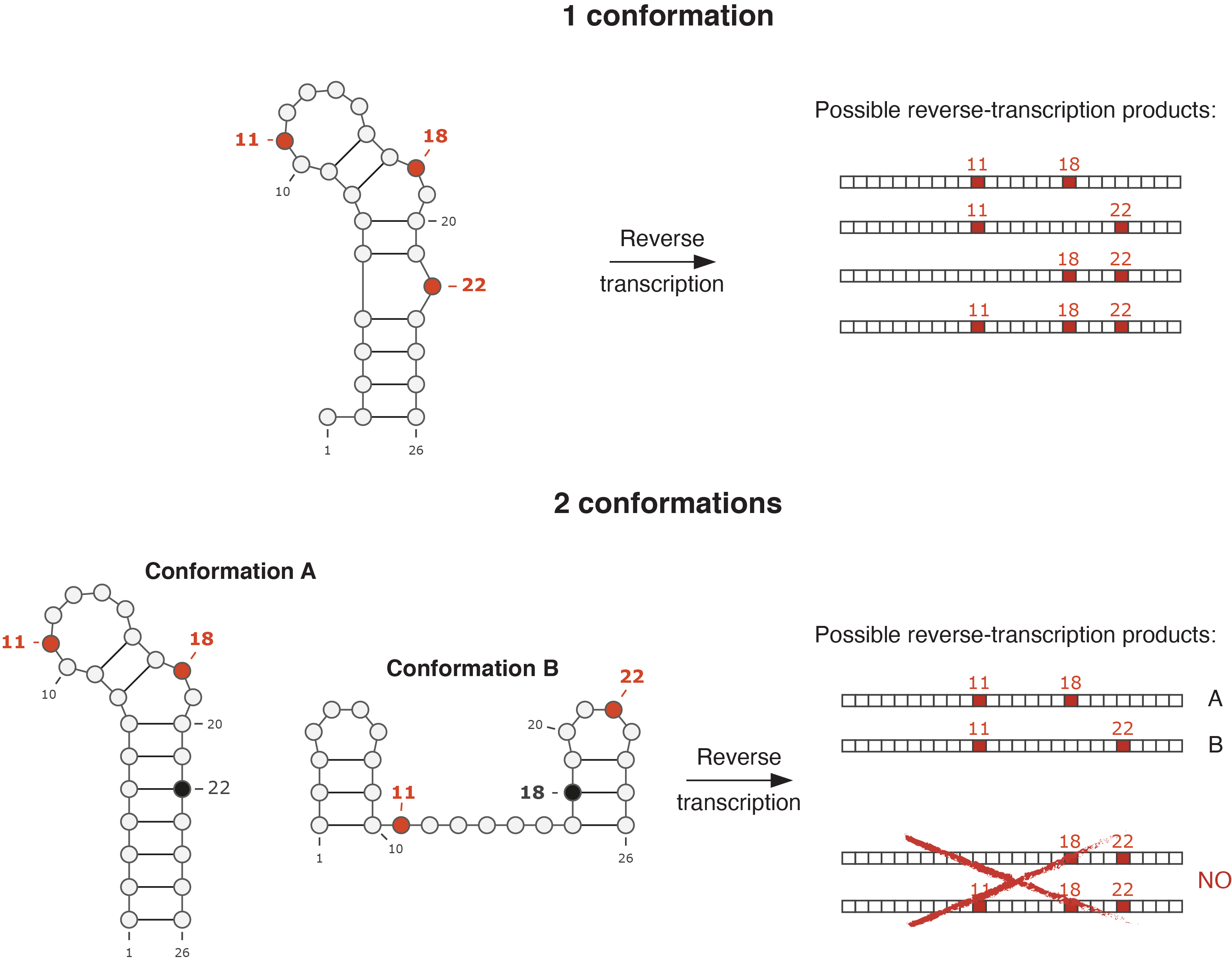
Example of RNA probing by mutational profiling. In one case, the RNA of interest folds into a single conformation. As bases 11, 18 and 22 are all single-stranded, they will all be recorded as mutations following reverse transcription, in the resulting DNA molecules. Therefore, all the possible combinations of mutations will be observed (11-18, 11-22, 18-22, 11-18-22). In the other case, the RNA folds into two mutually-exclusive conformations. Both conformations have base 11 single-stranded, but they differ in the configuration of bases 18 and 22. In this case, only two possible combinations of mutations will be observed, particularly 11-18 and 11-22, while no DNA molecule will carry simultaneous mutations on bases 18 and 22.
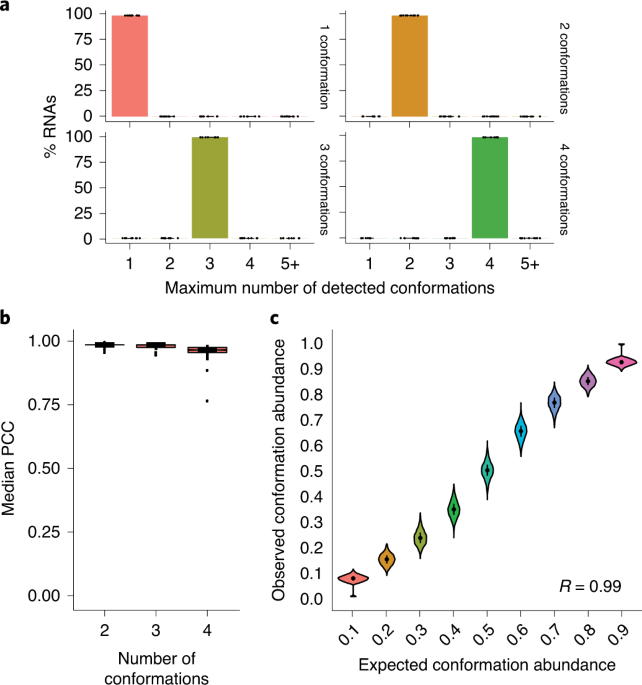
This was the starting point for the development of DRACO (Deconvolution of RNA Alternative COnformations). It took us nearly three years to develop a computational method robust and fast enough to be able to efficiently analyze the huge amounts of data generated in nowadays RNA probing experiments, and to enable the analysis of long RNA molecules as well. In our study, we decided to apply DRACO to the characterization of the RNA genome of the SARS-CoV-2 virus. This analysis revealed the presence of several key regions of the viral genome that can form (at least) two mutually-exclusive conformations. Although at this stage we cannot make any guess on the putative function of these potential structural switches, the conservation observed for some of them across several representatives of the coronavirus family hints at their functional relevance. Future studies will hopefully help us elucidating their function.

Schematic representation of the SARS-CoV-2 RNA genome. Genes are indicated as arrows. DRACO identified structurally-dynamic regions, and as such able to switch between alternative mutually-exclusive conformations. These include two important viral regulatory regions (marked in purple).
To learn more about DRACO, here is the link to the full article.
DRACO is freely available under GPLv3 license from GitHub.
Follow the Topic
-
Nature Methods
This journal is a forum for the publication of novel methods and significant improvements to tried-and-tested basic research techniques in the life sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Methods development in Cryo-ET and in situ structural determination
Publishing Model: Hybrid
Deadline: Oct 30, 2026


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in