
Chromosomal abnormalities are a hallmark of leukaemia, defining specific disease subtypes and correlating with clinical outcomes. Despite being a disease of adulthood, acute myeloid leukaemia (AML) can affect children, with a peak of incidence between the ages of 0 and 2 years (1). Interestingly, specific genetic abnormalities are only encountered in this age group and are totally absent from older children and adults (2). Over the years, increasingly in-depth analyses of leukaemia patient samples of varying ages have uncovered distinct features including transcriptional profiles, mutational landscape, and oncogenic mechanisms, which render each subtype a unique biological entity (3). Nevertheless, targeted therapies for infant-specific cytogenetic subtypes are lacking, with relapse, therapy-related toxicity, and mortality remaining major clinical challenges.
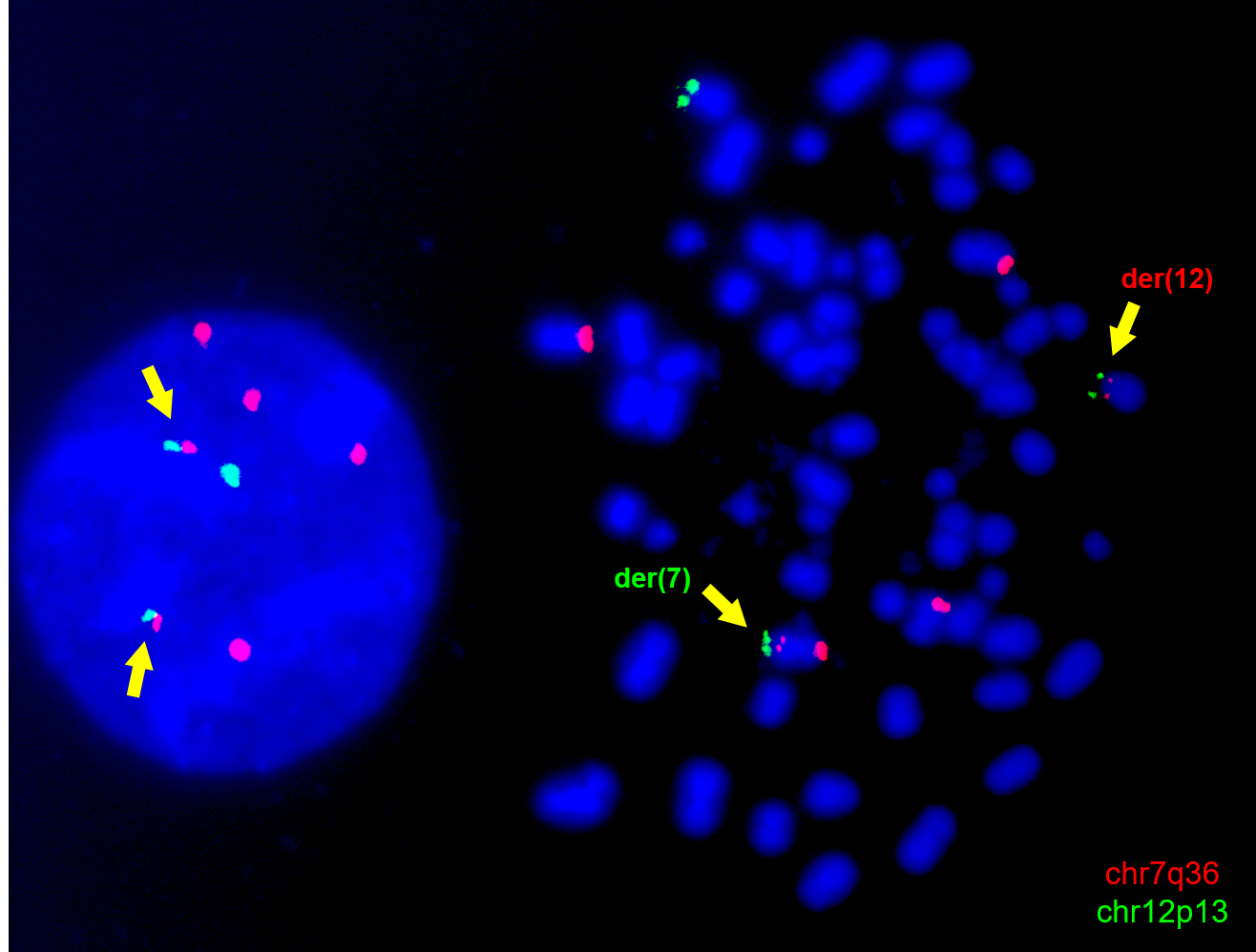
A particular focus of our laboratory has been the chromosomal translocation t(7;12)(q36;p13), which represents up to one third of infant AML cases below the age of 2 (Figure 1A). Since its discovery, the biology of t(7;12) has remained elusive, with the only known molecular consequence being the overexpression of the embryonic gene MNX1. Moreover, patients harbouring this translocation have notoriously poor clinical outcomes, which require research efforts to unravel its biology and tackle novel therapeutic interventions. However, a major constraint of research into t(7;12)-associated infant leukaemia has been the lack of research models (4).
The advent of genome editing technologies, first and foremost CRISPR/Cas9, has revolutionised systems biology, allowing the creation of increasingly accurate genetic modifications that were not possible before. In addition to disrupting or correcting individual genes, the CRISPR system opened a wealth of possibilities owing to the ability to generate chromosomal rearrangements (5). We decided to harness CRISPR/Cas9 to generate the t(7;12) translocation in vitro, in order to address the need for a research model (Figure 1B-C).
 in vitro.")
 in vitro.")
Using the well characterised and immortalised K562 leukaemia cell line allowed us to produce a stable tool that could retain the rearrangement and be propagated. Despite being already transformed, the translocation produced highly evident morphological changes linked to the differentiation potential of the cell line, which corresponded to the clinical manifestations of t(7;12) as poorly differentiated AML (Figure 1D). Interestingly, we originally attempted to edit other leukaemia lines (such as Jurkat), which failed to retain the translocation, possibly hinting at the requirement for a specific cellular or developmental background. In fact, K562 cells possess a more embryonic-like background, in virtue of their highly immature differentiation state and the production of foetal haemoglobins.
We further explored the age specificity of t(7;12) by introducing the translocation in healthy CD34+ adult haematopoietic progenitors (HSCPs). The translocation gradually disappeared while in culture and did not provide any survival or proliferative advantage (Figure 1E). In the attempt to provide a microenvironment-like support, CD34+t(7;12) HSCPs were co-cultured with MS5 stroma, but also failed to expand the t(7;12)-harbouring clones. As already reported by others (6,7), the overexpression of MNX1 lacks the ability to transform adult haematopoietic cells.
Nonetheless, the K562-t(7;12) model proved useful in recapitulating key features of t(7;12) AML patients. Based on the observations from Ballabio et al. (8) in t(7;12) patient samples, we were able to prove that the introduction of the translocation caused an altered genomic positioning of the affected loci in interphase nuclei. As seen in patients, the portion of the translocated chromosome 12 containing the MNX1 gene was repositioned towards the nuclear interior, a transcriptionally active compartment, which coincides with its ectopic overexpression. The upregulation of MNX1 in our model, which is a key feature of t(7;12) AML, was particularly remarkable for us, due to the pre-existing high expression of this gene in K562.
We also performed RNA sequencing to identify gene expression changes brought about by the translocation. Despite limited high-throughput transcriptomic analyses of t(7;12) patients, we made use of publicly available RNA sequencing and microarray experiments from the public database TARGET to extract gene expression signatures for t(7;12). Our K562-t(7;12) model recapitulated these signatures compared to the unedited K562 control, which further established that the dysregulation of these genes was linked to the generation of the rearrangement (Figure 1F-G). In addition, we observed an enrichment in cell adhesion processes, which have also been described in t(7;12) patients and linked to the disruption of haematopoietic niche interactions as pathogenetic mechanism (7).
We foresee that our engineered K562 model will help uncover transcriptional and mechanistic features of t(7;12) AML, thanks to its ease in manipulation as a research tool and unlimited lifespan. Our results further reinforced the notion that cellular / developmental background is an important determinant for the effects of t(7;12). In conclusion, our investigation on leukaemogenesis associated with t(7;12) continues, with a clinically-relevant model to faithfully phenocopy this particular subtype of AML.
References
(1) Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer Statistics, 2021. CA Cancer J Clin 2021 Jan;71(1):7-33.
(2) Creutzig U, van den Heuvel-Eibrink MM, Gibson B, Dworzak MN, Adachi S, de Bont E, et al. Diagnosis and management of acute myeloid leukemia in children and adolescents: recommendations from an international expert panel. Blood 2012 Oct 18;120(16):3187-3205.
(3) Bolouri H, Farrar JE, Triche Jr T, Ries RE, Lim EL, Alonzo TA, et al. The molecular landscape of pediatric acute myeloid leukemia reveals recurrent structural alterations and age-specific mutational interactions. Nat Med 2018;24(1):103.
(4) Tosi S, Kamel YM, Owoka T, Federico C, Truong TH, Saccone S. Paediatric acute myeloid leukaemia with the t (7; 12)(q36; p13) rearrangement: a review of the biological and clinical management aspects. Biomarker research 2015;3(1):1.
(5) Torres R, Martin M, Garcia A, Cigudosa JC, Ramirez J, Rodriguez-Perales S. Engineering human tumour-associated chromosomal translocations with the RNA-guided CRISPR–Cas9 system. Nature communications 2014;5:3964.
(6) Ingenhag D, Reister S, Auer F, Bhatia S, Wildenhain S, Picard D, et al. The homeobox transcription factor HB9 induces senescence and blocks differentiation in hematopoietic stem and progenitor cells. Haematologica 2019 Jan;104(1):35-46.
(7) Wildenhain S, Ruckert C, Röttgers S, Harbott J, Ludwig W, Schuster F, et al. Expression of cell–cell interacting genes distinguishes HLXB9/TEL from MLL-positive childhood acute myeloid leukemia. Leukemia 2010;24(9):1657.
(8) Ballabio E, Cantarella C, Federico C, Di Mare P, Hall G, Harbott J, et al. Ectopic expression of the HLXB9 gene is associated with an altered nuclear position in t (7; 12) leukaemias. Leukemia 2009;23(6):1179-1183.
Follow the Topic
-
Oncogenesis
A peer-reviewed open access online journal that publishes articles exploring mechanistic insight and molecular basis of cancer and related phenomena. It seeks to promote diverse and integrated areas of molecular biology, cell biology, oncology, and genetics.

Related Collections
With Collections, you can get published faster and increase your visibility.
Metabolic Reprogramming in Cancer
The field of cancer metabolism has expanded rapidly, revealing how metabolic reprogramming drives tumour progression, therapy resistance, and cellular adaptation. Beyond local interactions, accumulating evidence shows that circulating immune cells and blood‑borne factors—including metabolites, cytokines, and extracellular vesicles—actively influence tumour metabolic states. These systemic signals interact with tumour‑resident cells to shape metabolic plasticity, modulate survival pathways, and affect treatment responses.
Within the tumour microenvironment itself, metabolic interplay between cancer cells, immune cells, and stromal components remains a central determinant of tumour behaviour. Altered glycolytic flux, lactate‑driven microenvironmental changes, and cancer stem cell–associated metabolic adaptations contribute to tumour aggressiveness, immune evasion, and resistance mechanisms. Together, these local and systemic dimensions provide an integrated view of how metabolism underpins cancer progression.
This collection supports United Nations SDG 3: Good Health & Well-Being.
Topics of interest include:
- metabolic interactions within the tumour microenvironment
- cancer stem cell metabolism
- glycolytic pathways in tumour promotion
- tumour–immune cell metabolic interplay (local and systemic)
- interplay between circulating blood cells and tumour metabolism
- drug resistance and metabolic shifts
- protein expression in metabolic reprogramming
- role of transcription factors in metabolism
- signal transduction pathways affecting cancer metabolism
Publishing Model: Open Access
Deadline: Dec 31, 2026



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in