Exploiting the temporal dimension of hydrogen-deuterium exchange mass-spectrometry to localise protein-protein interactions
Published in Chemistry
Determining a protein's structure and conformational dynamics is key to understanding its function. One approach to explore this experimentally is hydrogen-deuterium exchange coupled to bottom-up mass-spectrometry (HDX-MS). By examining which peptides have altered exchange rates due to a protein-protein interaction, we can spatially localise interaction sites of those proteins and potentially the conformational change induced by that interaction. This experiment is performed by making mass-spectrometric measurements over a time-course of exposure of the protein, or proteins, to heavy water. Deuterium in the heavy water exchanges with hydrogen in the protein backbone and this uptake of deuterium is quantified by mass-spectrometry measurements of peptides within that protein. The rate of incorporation for each residue is determined by the protein's structure and conformational ensemble. HDX-MS is a popular technique because it is simple, yet, powerful. Few methods offer both the possibility of determining whether an interaction is occurring and the site of that interaction. When two proteins interact they form new hydrogen bonds, which slows the rates of exchange and we can measure this using MS. Potentially, rates of exchange can change elsewhere on the protein if the interaction biases the conformational ensemble of that protein. In an ideal scenario, those slowing of rates would be clearly visible. However, as with all experiments, technical variability can corrupt the measurements.
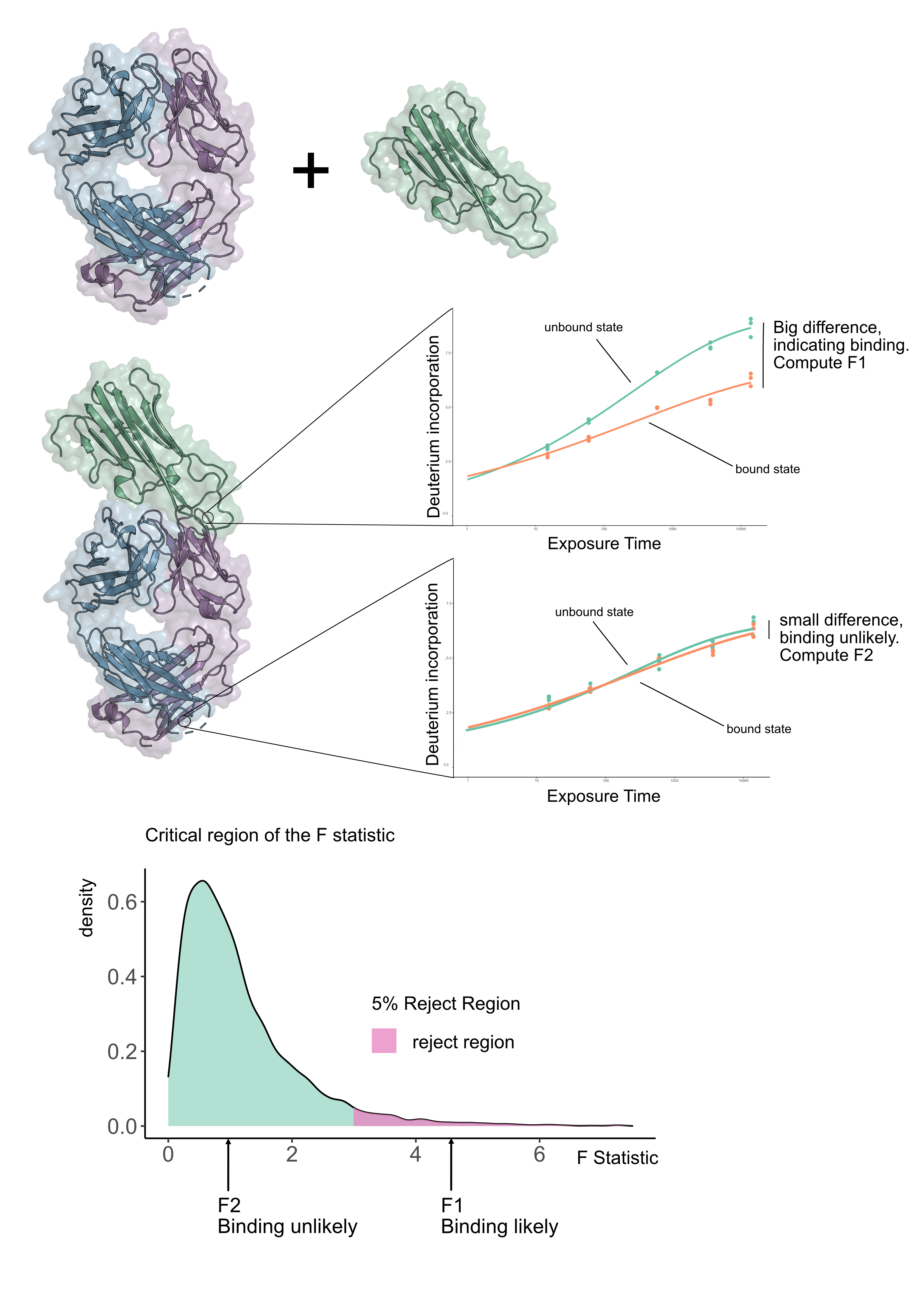
Suppose we have an antibody in its unbound form and in a form bound to an antigen (see Figure 1), we want to know where the antibody actually binds the antigen. We refer to this as epitope mapping. We collected data for a number of antibodies for HOIP-RBR, a regulator of inflammation, and sought to determine the epitopes in a systematic way using HDX-MS. We became frustrated because standard statistical methods gave inconclusive results. However none of these approaches used the the fact that HDX-MS is measured as a time-series. We rationalised that if we exploited the time dimension of HDX-MS data, we could make a more powerful statistical test. The idea hinges on fitting curves to the HDX-MS data and seeing how well they explain the variability in the data. If we fitted a curve for each condition for each peptide then if there is a difference between the kinetics then the curves would be sufficiently far apart. The next question is, how do we quantify whether these curves are sufficiently far apart? The answer is to fit a null model, which is a curve that does not know if we are in the unbound or bound state. The alternative model is one where independent curves are allowed for the bound and unbound states. To see which model was better, we quantified how well they explained the data using an F-statistic (see Figure 1). The size of the F-statistic is interpreted against the appropriate F-distribution (which is determined by the number of parameters in the model) and computing a p-value from it (see Figure 1).
We quickly realised that our approach led us to a powerful and reliable method for HDX-MS data analysis. To check that our method was working well, we examined experiments where there should be no differences. We found that our method only made incorrect assertions a very small number of times. Returning to our antigen-antibody experiment, we found that we were able to localise a number of epitopes in a statistically rigorous and sound way, where previously only qualitative inspection has been possible. We believe this will aid in therapeutic design because we can accurately and confidently identify the epitope.
See the paper: https://www.nature.com/articles/s42003-022-03517-3
Follow the Topic
-
Communications Biology
An open access journal from Nature Portfolio publishing high-quality research, reviews and commentary in all areas of the biological sciences, representing significant advances and bringing new biological insight to a specialized area of research.

Related Collections
With Collections, you can get published faster and increase your visibility.
Artificial Intelligence Methodology in Structural Biology
Publishing Model: Hybrid
Deadline: Nov 30, 2026
Healthy Aging
Publishing Model: Open Access
Deadline: Dec 31, 2026


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in