FEAST: deciphering the origins of the microbiome
Published in Protocols & Methods

This post was written by Liat Shenhav and Eran Halperin
What is microbial source tracking and why this is important?
Knowledge of the diverse functions and distributions of microbial life has rapidly increased due to an extraordinary expansion of microbiome data repositories like the “Earth Microbiome Project”. Such rich datasets provide the opportunity to study the relationships between the abundance profiles of bacteria in different habitats. Nonetheless, one critical challenge in analyzing microbiome communities is due to their composition; each community is typically a mixture of several source environments, as a result of natural processes of microbial colonization or the presence of contaminants. To account for this structure and unravel the origins of these complex microbial communities, methods for “microbial source tracking” have been proposed in the past decade. These methods quantify the fraction, or proportion, of different microbial samples (sources) in a target microbial community (sink).
How did we start working on it?
Microbial source tracking has been widely used by the community after a 2011 Nature Methods publication by Rob Knight’s group proposed a method that deals with this problem while utilizing the structure of the entire microbial community. Their method has been widely applied over the past 8 years, and it has been extremely useful for tracking the formation of microbial communities. However, the nature of the data that existed in 2011 was very different from the data that exists today, in terms of scale, quality, and availability. Indeed, we realized that some source tracking tasks simply cannot be performed using existing tools when applied to modern data. Particularly, in 2017, we were collaborating with Itzik Mizrahi’s group from Ben-Gurion University (in Israel) on a project that aimed to characterize the development of microbial communities in the rumen. Their experiment required the analysis of a very large dataset, and as a consequence, computational efficiency was a crucial component. Specifically, an integral part of the analysis included the application of microbial source tracking methods in order to decipher the origins of the rumen microbiome.
When we attempted to apply existing source tracking methods to their dataset we found that due to its size, these methods either crashed or required many months of processing on a large computer cluster. We then decided to dive deeper into the details of the original source tracking methods and realized that this problem is actually very similar to ancestry inference from human genetic data, a problem that our group has been studying for many years. Figuring out how a microbial community is formed, is similar to figuring out how many immigrants came to a specific country from other countries. For example, the major immigration waves to Brazil are from Europe, Asia, and West Africa. Quantifying the proportion of European, African and Asian ancestry in the Brazilian population is a classical problem in statistical genetics.
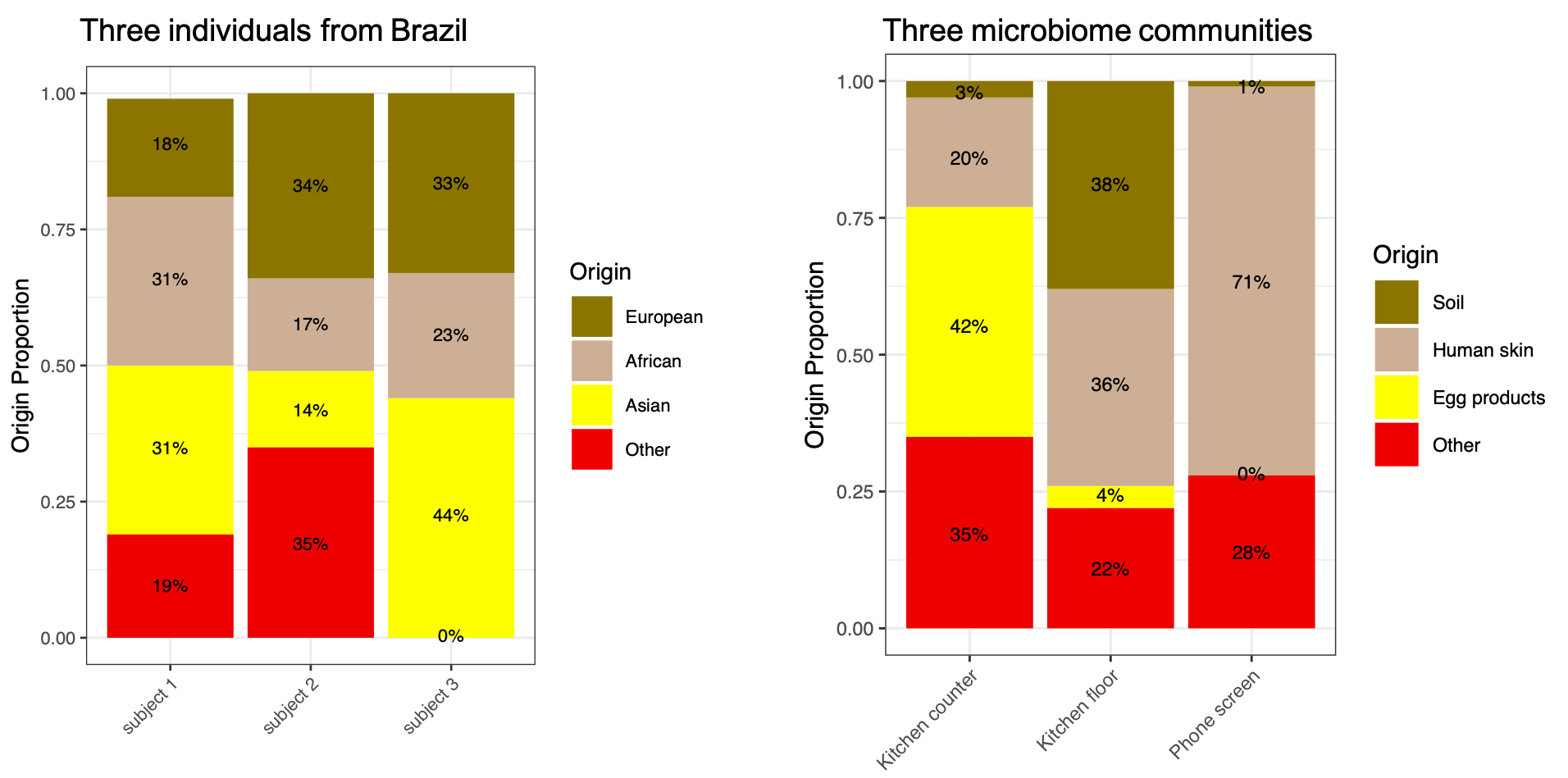
Figure 1: Just like we can estimate the origins of individuals from Brazil (Europe, Asia, Africa, and other), we can estimate the origins of microbial communities (soil, human skin, egg products and other).
Notably, the resemblance does not end here. In statistical genetics, a similar improvement pattern occurred. Specifically, this mixture problem has been initially addressed using a Bayesian approach in a software tool called STRUCTURE by Jonathan Pritchard and colleagues. However, this method was not efficient enough to process large datasets that were generated just a few years later. Therefore, we and others came up with more efficient methods that enabled rapid inference of population structure - a mechanism extensively used by geneticists. ‘Armed’ with this insight, we started structuring the FEAST probabilistic model with the hope of replicating the success story of ancestry inference in statistical genetics.
What can we achieve using FEAST?
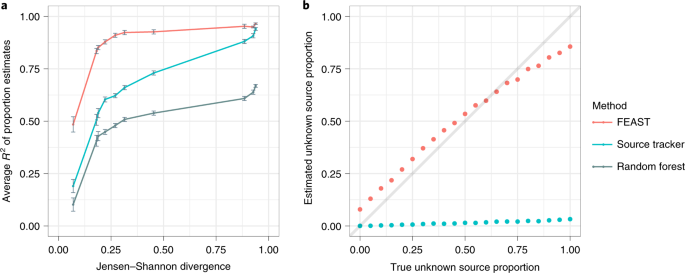
We then worked very hard on the nuts and bolts of the model, resulting in a method that reduced the running time of state-of-the-art methods by a factor of 30-300. Consequently, FEAST can simultaneously estimate thousands of potential source environments on the order of minutes to hours. To our surprise, in addition to the gain in speed, we also found that FEAST is significantly more accurate than previous methods across many different scenarios.
After multiple data-driven simulation studies, we were convinced that our method provides a considerable improvement in terms of speed and accuracy. We next established the biological utility of FEAST in two different contexts. First, we used FEAST on a time-series dataset of 98 mothers and their infants along the first year of life, in order to track the formation of the gut microbial community. Using FEAST, we quantitatively reaffirmed that the gut microbiota of infants delivered by C-section showed significantly less resemblance to their mothers’ compared to vaginally-delivered infants (initially demonstrated by Backhed and colleagues in 2015). Second, we used FEAST as a metric of similarity, a novel application for microbial source tracking. Using FEAST, we were able to differentiate between the gut microbiome of ICU patients experiencing dysbiosis and that of healthy controls. The results from FEAST show that the gut microbiome of healthy adults demonstrates a greater resemblance to other healthy gut communities than to those of patients experiencing dysbiosis.
Our hope is that FEAST will become routinely used in the pipeline for the analysis of microbiome data. For this reason, we released a user-friendly R package (https://github.com/cozygene/FEAST), and we are in the process of adding FEAST to QIIME II, an open source bioinformatics tool that is widely used for microbiome analysis. We anticipate that using FEAST important biological insights will be revealed in the fascinating world of the microbiome.
Follow the Topic
-
Nature Methods
This journal is a forum for the publication of novel methods and significant improvements to tried-and-tested basic research techniques in the life sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Methods development in Cryo-ET and in situ structural determination
Publishing Model: Hybrid
Deadline: Oct 30, 2026


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in