Fixing the Broken Promise: FDR-Controlled Framework for Microbial Biomarker Detection
Published in Microbiology, Protocols & Methods, and Computational Sciences
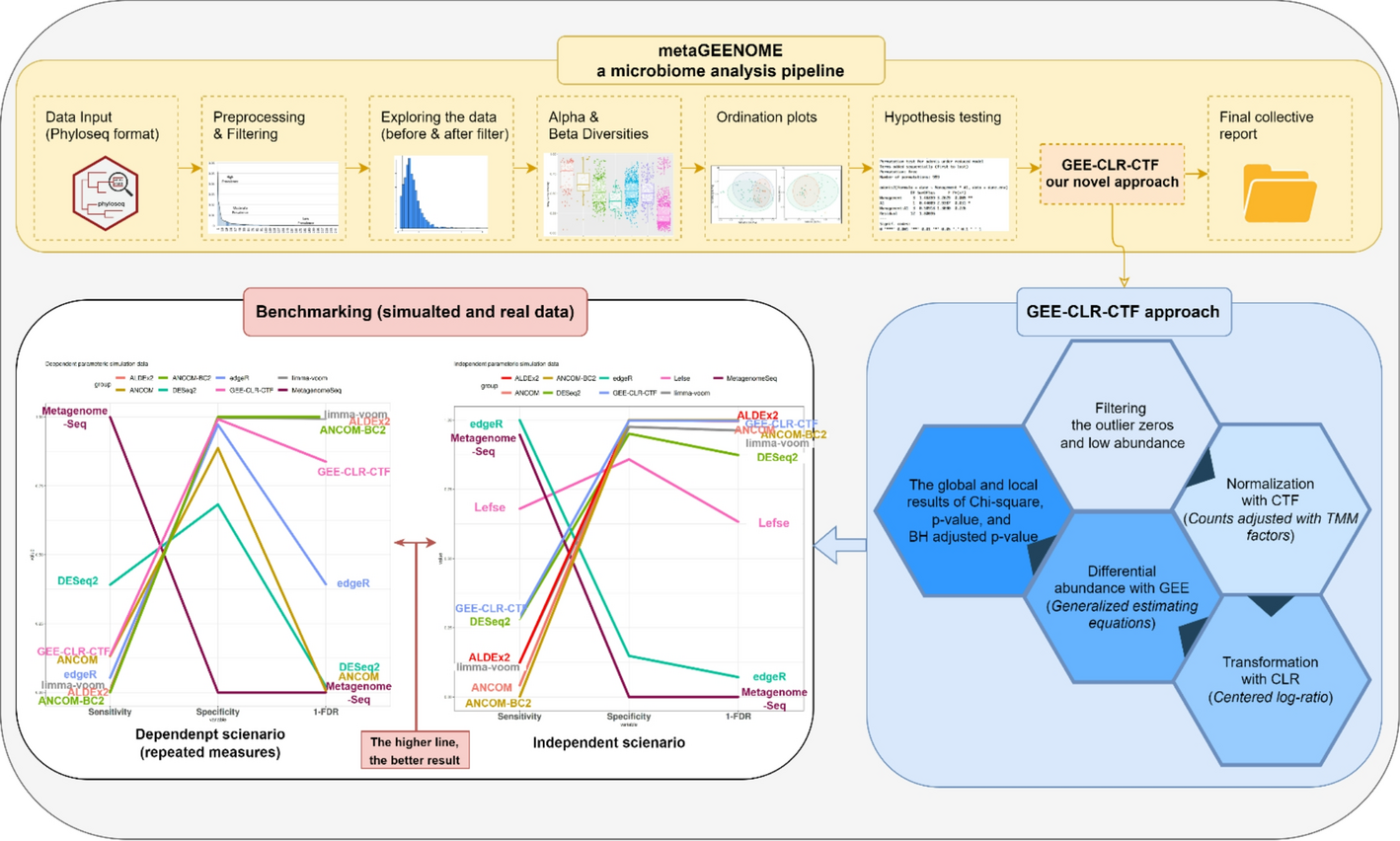

Microbiome research has rapidly advanced thanks to breakthroughs in DNA sequencing, enabling scientists to explore the microbial ecosystems that inhabit our bodies, environments, and foods. One of the most important questions in this field is: Which microbes are associated with specific conditions—like diseases, treatments, or environmental changes? The ability to detect these microbial “biomarkers” reliably and reproducibly is essential for progress in medicine, agriculture, and public health. However, despite the popularity of microbial differential abundance (DA) analysis tools, a troubling issue has emerged—many widely used methods struggle to balance sensitivity (the ability to find true signals) with false discovery control (avoiding spurious ones). This problem is especially pronounced in longitudinal studies, where samples are collected over time from the same individuals.
This challenge was highlighted by several previous reviews, including a landmark paper by Hawinkel et al. (2019), which showed that commonly used tools often fail to control the false discovery rate (FDR), raising questions about the reproducibility of microbiome findings. More recently, Pelto et al. (2024) emphasized the need for statistical methods that are not only accurate but also robust to data heterogeneity and repeated measurements. These concerns resonated with our own experiences analyzing microbiome data, where tools would often report conflicting or inconsistent results depending on the dataset structure or preprocessing steps.
The "Broken Promise"
At the heart of the problem lies a “broken promise”: most existing DA tools promise to identify important microbial shifts while maintaining rigorous statistical standards. In practice, they often fall short of this balance. Tools such as DESeq2, edgeR, MetagenomeSeq, and LefSe can be very sensitive—detecting many differences—but they frequently fail to control FDR, leading to false positives. On the other hand, methods like ALDEx2, ANCOM, and ANCOM-BC2 maintain stricter control over false discoveries, but at the cost of missing many real microbial associations. This trade-off forces researchers to choose between power and precision, when ideally both should be achievable.
Our goal in this study was to “fix the broken promise” by developing a statistically sound framework that performs well across cross-sectional and longitudinal designs. We wanted a method that could offer robust FDR control while still detecting meaningful microbial signals—particularly in real-world datasets, which often feature complex structures such as compositionality, sparsity, inter-taxa correlations, and repeated measurements.
A New Framework: GEE‑CLR‑CTF
To meet this challenge, we developed a new analytical framework that combines three key components:
-
GEE (Generalized Estimating Equations):
GEE is a well-established statistical method that accounts for correlation within repeated measurements. Unlike standard models that assume independence across samples, GEE recognizes that samples from the same individual (or environment) may be inherently related. This makes it ideal for analyzing longitudinal microbiome data or studies with repeated sampling designs. -
CLR (Centered Log-Ratio) Transformation:
Microbiome data are compositional, meaning only the relative abundances of taxa are observed. CLR transformation is a common and theoretically sound way to address this issue by transforming the data into a scale where meaningful statistical comparisons can be made. -
CTF Normalization (Counts adjusted with Trimmed Mean of M-values):
Before applying CLR, we normalize the raw count data to mitigate differences in sequencing depth and library size. CTF adapts techniques from RNA-seq normalization (like TMM) to better handle zero-inflation and extreme values, which are common in microbial datasets.
Together, these steps form the GEE‑CLR‑CTF framework—a novel and integrated pipeline tailored specifically for the statistical challenges of microbiome studies.
metaGEENOME: Turning the Framework into a Tool
To make our method accessible to the broader scientific community, we developed an open-source R package called metaGEENOME, available here: https://github.com/M-Mysara/metaGEENOME. This package implements the full pipeline, including:
-
Preprocessing of microbial count tables
-
Zero and outlier filtering
-
CTF normalization and CLR transformation
-
GEE-based modeling
-
Global and local hypothesis testing
-
Result visualization and export
The tool is designed to work with standard microbiome data formats and provides easy integration with other R-based workflows, making it practical for researchers with varied levels of programming experience.
Benchmarking and Validation
We rigorously benchmarked our method using both simulated and real datasets. In the simulated studies, we designed scenarios that mimic real microbiome data complexities, including high-dimensional feature spaces, uneven sequencing depth, inter-taxa dependencies, and within-subject correlations. Our framework consistently outperformed popular DA tools on key metrics:
-
False Discovery Rate (FDR):
Our method achieved strict control of FDR, with values near or below 0.5% in cross-sectional settings and below 15% in longitudinal scenarios. -
Specificity:
The framework also delivered high specificity (≥99.7%), ensuring that most of the detected signals are true.
In real-world case studies, including publicly available longitudinal microbiome datasets, metaGEENOME demonstrated consistent performance, identifying biologically meaningful microbial biomarkers and replicating findings across dataset partitions. This addresses concerns raised by Pelto et al. (2024) about reproducibility and cross-study stability.
Addressing the Core Questions
In their 2019 paper, Hawinkel et al. asked whether commonly used methods truly control the false discovery rate in microbiome analysis—and concluded that many do not. Our work answers this concern directly: by building a method with robust statistical underpinnings and validating it across realistic conditions, we demonstrate that it is possible to control FDR without giving up sensitivity.
Similarly, Pelto et al. emphasized the need for tools that are sensitive to data structure, particularly longitudinal and repeated-measure designs. Our use of GEE addresses this head-on. While most DA tools assume independence between samples, GEE appropriately models intra-subject correlations, filling a gap in current practice and expanding the applicability of DA analysis to a broader range of study designs.
Broader Impact
We believe the GEE‑CLR‑CTF framework and the metaGEENOME package represent an important step forward for microbiome research. By providing a more reliable and reproducible method for differential abundance analysis, we hope to empower researchers to draw more confident conclusions from their data. This is particularly critical as microbiome studies grow in scale and importance, with applications in precision medicine, environmental monitoring, nutrition, and beyond.
Moreover, because our approach is open source, reproducible, and designed to be compatible with standard microbiome workflows, it lowers the barrier to adoption for research teams across disciplines. We also see opportunities for future extensions of this work, including adaptation to other types of omics data, integration with multi-omics pipelines, and further improvements in computational efficiency.
Access the Work
The full paper is available open-access here:
📄 https://rdcu.be/exfIj
The metaGEENOME R package is available on GitHub:
🛠️ https://github.com/M-Mysara/metaGEENOME
Follow the Topic
-
BMC Bioinformatics
This is an open access, peer-reviewed journal that considers articles describing novel computational algorithms and software, models and tools, including statistical methods, machine learning and artificial intelligence, as well as systems biology.

Related Collections
With Collections, you can get published faster and increase your visibility.
Computational methods in paleobiology
BMC Bioinformatics is welcoming submissions to our Collection on Computational methods in paleobiology.
The interdisciplinary field of paleobiology enables researchers to reconstruct evolutionary histories, model population dynamics, and explore the multifactorial influences that shaped life in the past. Paleobiology includes, but is not limited to, macrofossils, microfossils, and ancient proteins, DNA and RNA from both fossils and environmental sources. For instance, with the rapid growth of paleogenomic datasets, novel computational approaches are essential for extracting insights from this fragmented ancient data.
This Collection welcomes submissions on the development of new computational and/or statistical approaches for paleobiology. We encourage contributions that highlight innovative methods for analyzing ancient DNA, modeling evolutionary processes, integrating heterogeneous datasets, and visualizing temporal and spatial patterns.
All manuscripts submitted to this journal, including those submitted to collections and special issues, are assessed in line with our editorial policies and the journal’s peer-review process. Reviewers and editors are required to declare competing interests and can be excluded from the peer review process if a competing interest exists.
Publishing Model: Open Access
Deadline: Jan 23, 2027
Cell tracking
BMC Bioinformatics is welcoming submissions to our Collection on Cell Tracking.
Cell tracking is a technique used to monitor and analyze the movement and behavior of cells over time, allowing the study of cellular behaviors, dynamics, and interactions within various biological contexts. Advanced bioinformatics tools play a vital role in analyzing cell tracking data. They help identify cell movement patterns and understand their biological implications. These tools are particularly relevant when processing large datasets and when investigating cell cycles.
This Collection welcomes submissions on the development of new computational and/or statistical approaches for cell tracking. We encourage contributions detailing methods for detecting and characterizing cell movements to better understand cell migration and behavior.
All manuscripts submitted to this journal, including those submitted to collections and special issues, are assessed in line with our editorial policies and the journal’s peer-review process. Reviewers and editors are required to declare competing interests and can be excluded from the peer review process if a competing interest exists.
Publishing Model: Open Access
Deadline: Sep 23, 2026
Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in