Unmasking Cancer’s Microbial Allies: A Mega-Analysis of the Vaginal Microbiome in Cervical Cancer
Published in Microbiology

Introduction
Cervical cancer (CC) remains the fourth most common malignancy among women worldwide, with over 600,000 new cases and more than 340,000 deaths each year. Persistent infection with high-risk human papillomavirus (HPV) is the primary driver of CC development, yet infection alone is insufficient to cause malignancy. The majority of HPV infections resolve spontaneously, suggesting that additional biological factors modulate progression to high-grade lesions and invasive cancer.
Recent research has pointed to the vaginal microbiome as one such factor. A healthy vaginal ecosystem is typically dominated by Lactobacillus species, which maintain a low pH, produce antimicrobial compounds, and contribute to mucosal immune defense. In contrast, dysbiotic states—characterized by reduced Lactobacillus abundance and increased anaerobic diversity—have been associated with higher HPV persistence, chronic inflammation, and epithelial barrier disruption. These microenvironmental changes may facilitate viral integration, immune evasion, and carcinogenesis.
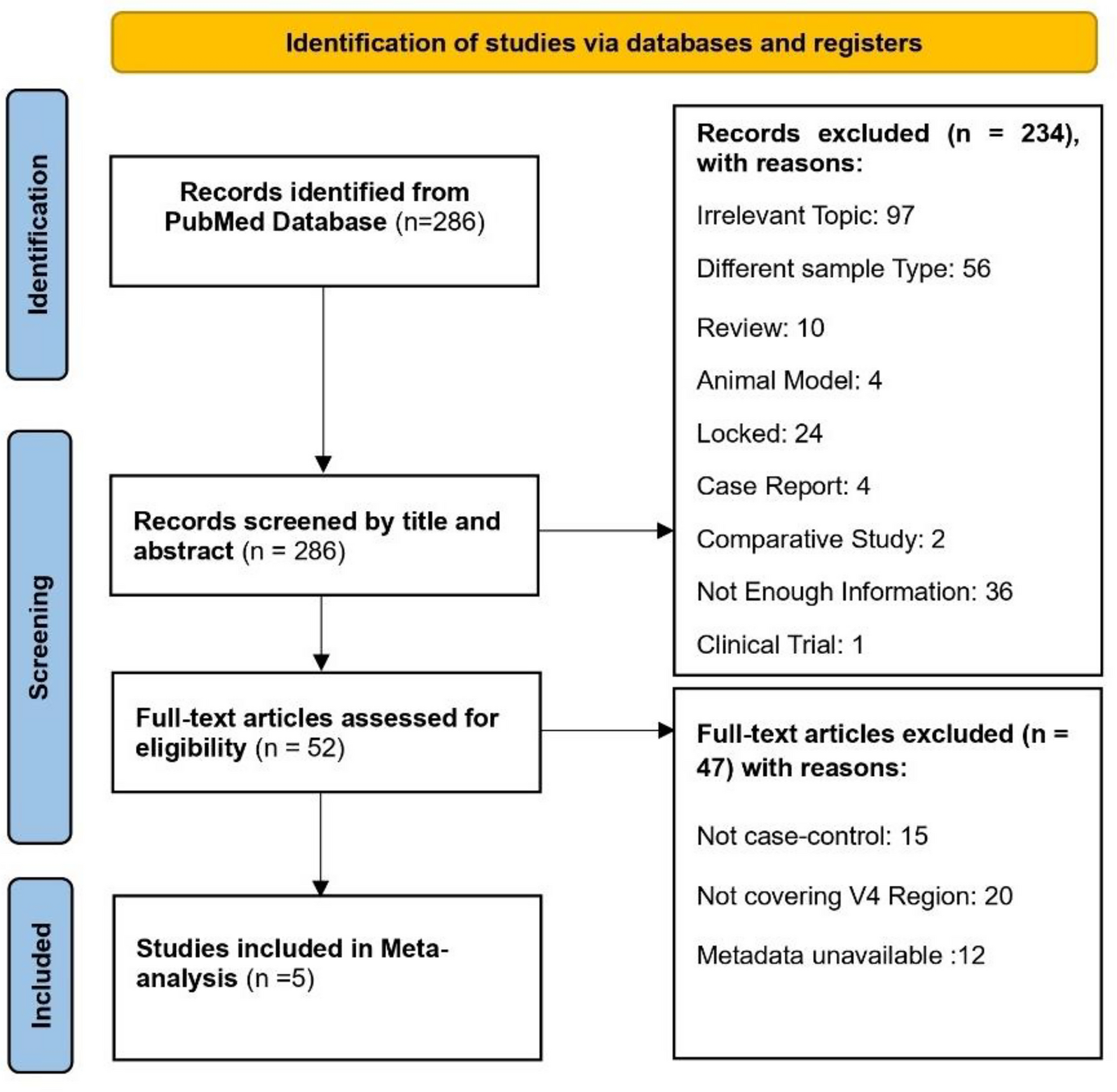
However, the literature on CC-associated microbiome shifts is fragmented. Studies vary in their sampling strategies, sequencing platforms, targeted 16S rRNA regions, and analytical approaches, leading to inconsistent and sometimes contradictory findings. Without harmonized data analysis, it is difficult to distinguish genuine biological patterns from methodological noise.
To overcome these challenges, we performed a compositionality-aware mega-analysis of all publicly available CC microbiome datasets meeting strict inclusion criteria. By reprocessing raw sequence data from multiple studies through a unified bioinformatic pipeline, we minimized technical bias and maximized comparability. Our goal was to identify reproducible microbial signatures and functional pathways linked to CC and its HPV-positive subsets—findings that could lay the groundwork for novel diagnostic and preventive strategies.
Key Findings
Our standardized analysis revealed a consistent shift in CC microbiota from a Lactobacillus-dominated community to a more diverse, anaerobe-rich profile. Alpha diversity was significantly higher in CC, and enriched taxa included Porphyromonas asaccharolytica, Campylobacter ureolyticus, Peptococcus niger, and Anaerococcus obesiensis. Concurrently, protective Lactobacillus species, particularly L. crispatus, were markedly depleted, especially in HPV-positive CC.
Functional predictions indicated enrichment in pathways related to fatty acid biosynthesis, oxidative phosphorylation, and altered amino acid metabolism—changes consistent with known cancer biology. Several of these pathways mirrored host transcriptomic profiles from independent CC datasets, pointing to possible microbial–host metabolic convergence.
Machine learning models trained on these microbial profiles achieved high predictive performance (up to 93% accuracy with XGBoost), underscoring the translational potential for microbiome-based diagnostics.
Future Directions
We faced limitations in geographic diversity, sample size, and clinical metadata. Our next steps involve expanding to more diverse populations, integrating shotgun metagenomics and metabolomics, and exploring causality through experimental models. Ultimately, we aim to extend this analytical framework to other female-related cancers, seeking shared microbial “fingerprints” and actionable biomarkers.
Final Remark
This first paper in our female cancer microbiome project establishes a reproducible foundation for studying microbial influences on HPV-driven cancers. By harmonizing data and revealing robust microbial and functional signatures, we open pathways for targeted diagnostics and prevention strategies—not only in cervical cancer, but across the spectrum of female malignancies. Read the full paper here: https://rdcu.be/eAuwO
Follow the Topic
-
Human Genomics
Human Genomics is a peer-reviewed, open access journal that focuses on the application of genomic analysis in all aspects of human health and disease, as well as genomic analysis of drug efficacy and safety, and comparative genomics.

Related Collections
With Collections, you can get published faster and increase your visibility.
Artificial Intelligence in Omics and Translational Research
This special Collection in Human Genomics focuses on the integration of artificial intelligence (AI), machine learning (ML), and deep learning methodologies with omics and multi-omics data, with a particular emphasis on human health and disease modeling. As AI technologies rapidly evolve, their potential to revolutionize omics research and precision medicine is - especially in understanding genetic variation in human populations and advancing disease modeling - is becoming increasingly clear. However, bridging the gap between omics experts and AI specialists remains crucial for fully harnessing this potential.
We invite manuscripts featuring original studies that utilize state-of-the-art AI and deep learning algorithms applied to clinical, population-based, or other human-focused omics and multi-omics datasets. Contributions should demonstrate clearly how these methodologies can identify biomarkers, elucidate complex disease mechanisms, and inform precision medicine strategies, presented in a way accessible to researchers without extensive computational backgrounds. Additionally, this Collection aims to inform AI and computer science communities about the types and availability of omics datasets, with a focus on human-centric applications, thereby promoting interdisciplinary collaborations. Submissions that provide practical guidance, review emerging AI methodologies, offer clear tutorials, or highlight successful interdisciplinary case studies are also highly encouraged.
Together, we seek to enhance AI literacy within the omics community and foster collaborative innovation, accelerating translational discoveries and advancing human genomic applications with tangible impact on public health. Topics of interest include but are not limited to:
• Original research applying deep learning algorithms to human-focused omics and multi-omics datasets
• Reviews highlighting the current state-of-the-art AI methodologies, including generative and foundational models, and their applications in human disease modeling
• Tutorials introducing deep and graph-based learning concepts tailored for omics researchers
• Descriptions and characterizations of publicly available omics datasets suitable for AI applications, with emphasis on clinical or population-based datasets
• Perspectives and case studies on interdisciplinary collaboration between omics scientists and AI specialists
We encourage clear, concise, and accessible writing for interdisciplinary readers to support greater integration across these rapidly advancing fields.
This Collection supports and amplifies research related to SDG 3, Good Health and Well-Being .
Publishing Model: Open Access
Deadline: Nov 15, 2026
Genomics of COVID-19: Molecular Mechanisms Going from Susceptibility to Severity of the Disease
The current COVID-19 pandemic has highlighted the importance of science and medicine, specifically public health, in our modern societies. Countries have taken different approaches to the pandemic. Science and medicine will play an important role in our way forward in tackling COVID-19. Specifically, genetics and genomics will be central in discovering variations in virus strains and their impact on patients’ outcome, the hosts’ ability to fend off the virus and the severity of disease in patients. Furthermore, the question of long-term immunity to COVID-19 may have a genetic and genomic basis which should be investigated. Some of these human genetics and genomics investigations will undoubtedly be suitable for publication in Human Genomics. We expressly welcome submissions of manuscripts on such subjects.
Publishing Model: Open Access
Deadline: Ongoing
Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in