Unlocking Reliable 16S rRNA Analysis: A Benchmarking Gold-Standard Ground Truth!
Published in Microbiology and Protocols & Methods
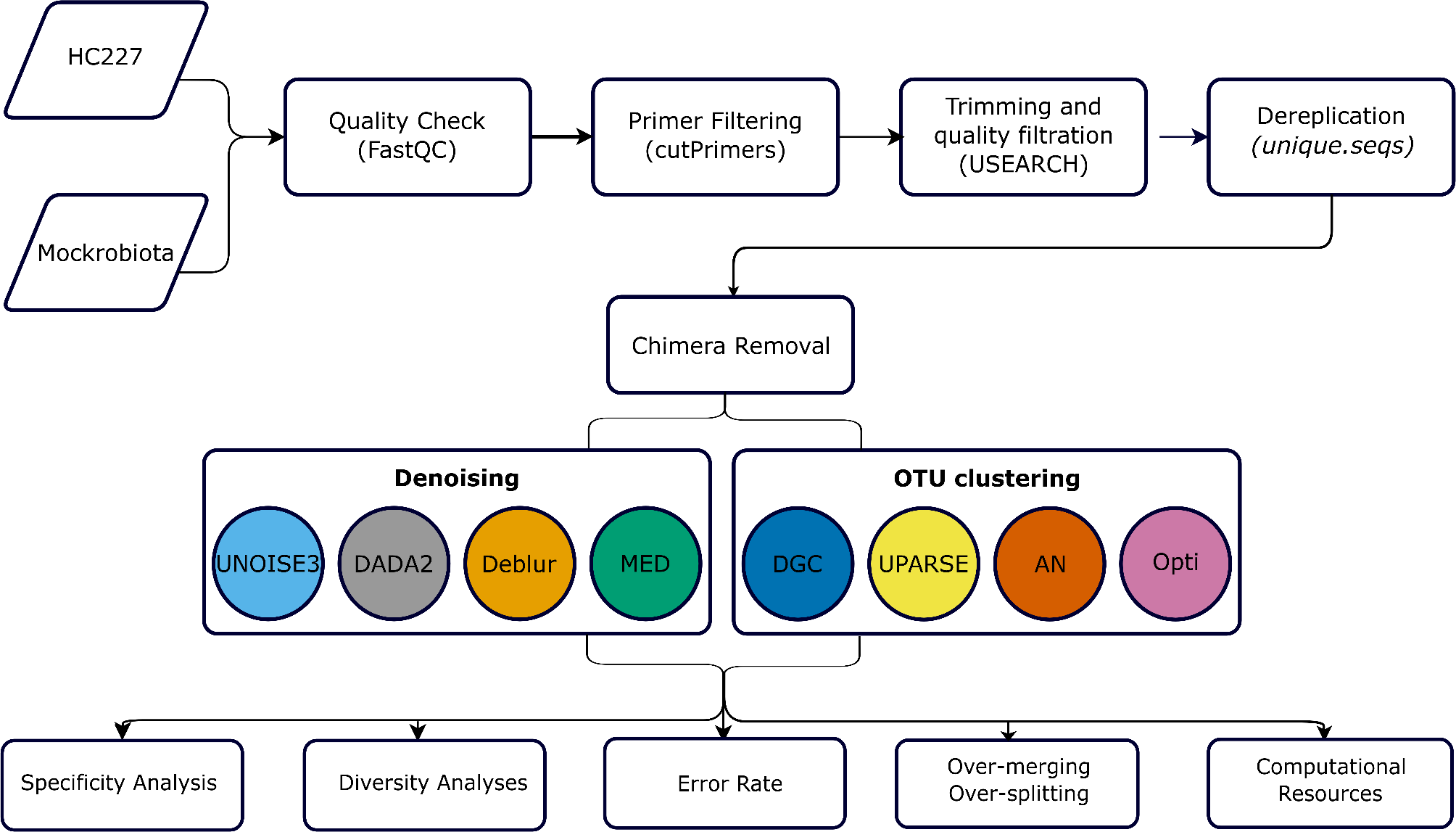

The analysis of 16S rRNA gene sequencing data involves several critical steps, including preprocessing, dereplication, chimera removal, and ultimately, clustering or denoising to infer biological sequences. To accurately assess the performance of each of these steps and ensure reliable results, the use of a complex mock community with a validated ground truth is essential for proper benchmarking. While large volumes of publicly available data exist and offer the advantage of being derived from real samples—unlike simulated data, which relies on prior assumptions—these datasets present a significant limitation: the true composition of the microbial communities is often unknown. This lack of a definitive ground truth poses a major challenge for comparative analyses, as it hampers our ability to rigorously evaluate the accuracy and effectiveness of clustering and denoising algorithms.
The mock community presented in this study comprises 235 bacterial strains representing 197 distinct species, providing a valuable and rigorous resource for the bioinformatics community. It offers an ideal framework for developers aiming to optimize their algorithms, as well as for analysts seeking to critically assess and benchmark existing 16S rRNA analysis pipelines. Notably, this same mock community has also been previously characterized at the shotgun metagenomic level by Gleb Goussarov, facilitating accurate metagenomic binning (see publication: https://link.springer.com/article/10.1186/s40793-022-00403-7). This dual availability at both amplicon and shotgun levels further enhances its utility as a comprehensive benchmarking standard for diverse microbial analysis workflow
In this study, we leveraged the complex mock community to conduct a head-to-head comparison of clustering and denoising approaches—specifically, Operational Taxonomic Units (OTUs) and Amplicon Sequence Variants (ASVs). This direct comparison allowed us to systematically highlight the strengths and limitations of each method. We believe that the robust design of our benchmarking framework, combined with the utilization of this complex mock community, provides a solid foundation for evaluating 16S rRNA analysis algorithms. Moreover, this framework offers a scalable model that could be extended to encompass entire pipeline comparisons in future studies.
Our comprehensive benchmarking framework, along with all datasets, detailed analyses, and key insights, is freely accessible https://environmentalmicrobiome.biomedcentral.com/articles/10.1186/s40793-025-00705-6.Additionally, the mock community dataset—available under accession number PRJNA975486—serves as a valuable resource for both bioinformatics algorithm development and rigorous performance evaluation.
Follow the Topic
-
Environmental Microbiome
Environmental Microbiome acknowledges the universal presence of microorganisms, which can be found across all environments on Earth, and is seeking submissions addressing the varied facets of environmental and applied microbiological research.

Related Collections
With Collections, you can get published faster and increase your visibility.
The global virome across changing ecosystems
Viruses are the most abundant biological entities on Earth, shaping microbial communities, driving biogeochemical cycles, and influencing their ecosystems in many ways. As environmental change accelerates (e.g. ocean warming, freshwater acidification, soil degradation, agricultural intensification, atmospheric shifts, and the emergence of space‑associated microbial habitats) the global virome is surely being affected – but how and to what extent remains largely unexplored.
This cross‑journal special Collection brings together research highlighting how viral diversity, ecology, function, and evolution are responding to our rapidly changing planet, maybe even impacting the change themselves and potentially acting as solutions to ameliorate some changes.
We would like to see submissions that go beyond describing the viral communities: we are looking for are studies that deepen our understanding of the roles viral communities play in our changing environments. Methodological advances (including metagenomics, multi‑omics, modelling, remote sensing, and experimental ecosystem approaches) are also encouraged. Through this Collection we would like to advance a holistic view of the planetary virome and its influence on life and the environment across our interconnected ecosystems.
Publishing Model: Open Access
Deadline: Mar 31, 2027
The Apple Microbiome
Microbiome and Environmental Microbiome are calling for submissions to our Collection on the Apple Microbiome.
With world apple production estimated at 84 million tons, the microbiome of the apple has significant implications for agriculture, food security, and human health. Understanding the complex interactions between apple plants and their associated microbial communities can lead to improved crop management strategies, enhanced fruit quality and longevity, and sustainable agricultural practices. Recent advances have highlighted the role of specific bacteria and fungi in promoting plant health and resilience against specific pathogens. Moreover, detailed profiling of these microbial communities, revealing their diversity and functional potential facilitate exciting future developments, such as the identification of beneficial microbial consortia for biocontrol and the formulation of tailored probiotic treatments for both plants and humans. By advancing our collective understanding in this area, we can work towards a more sustainable and resilient agricultural system.
Topics of interest include but are not limited to:
-Microbial diversity and function associated with apples
-Effects of soil health and rhizosphere interactions on apple production
-Impact of climate change on the apple microbiome
-Role of the apple microbiome in fruit quality
-Microbiome-driven strategies for disease resistance
This collection is open for submissions from all authors on the condition that the manuscript falls within both the scope of the collection and the journal it is submitted to.
All submissions in this collection undergo the relevant journal’s standard peer review process. Similarly, all manuscripts authored by a Guest Editor(s) will be handled by the Editor-in-Chief of the relevant journal. As an open access publication, participating journals levy an article processing fee (Microbiome, Environmental Microbiome). We recognize that many key stakeholders may not have access to such resources and are committed to supporting participation in this issue wherever resources are a barrier. For more information about what support may be available, please visit OA funding and support, or email OAfundingpolicy@springernature.com or the Editor-in-Chief of the journal where the article is being submitted.
Publishing Model: Open Access
Deadline: Sep 30, 2026
Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in