How macrophage interactions with apoptotic cells might contribute to parasite infection and pathology in Chagas disease?
Published in Microbiology
A long research way from the first discovery…
Chagas disease is considered a neglected infectious disease that is endemic in Latin America. Patients infected with the protozoan parasite Trypanosoma cruzi can develop a progressive cardiac illness that causes heart malfunction, heart failure and death. We still do not have vaccines or medicines that either prevent or treat the established disease, in part because the mechanisms of chronic pathology remain elusive.
The immune system is important to control but fails to eliminate parasite infection. Moreover, both the immune response and chronic infection cause damage to the affected tissues and organs. Our research focuses on the reasons why immune cells such as T lymphocytes and macrophages are insufficient to eradicate infection.
Three decades ago, I and my PhD adviser observed that T cells from T. cruzi-infected mice showed a defective proliferative response compared to T lymphocytes from healthy mice. Furthermore, T cells from infected mice started a cell death program (apoptosis) and thereby did not divide to produce new cells to fight infection.
By the end of the century, the clearance of apoptotic cells by macrophages was no longer considered a simple silent process that prevented the leakage of dangerous cell contents but also a suppressive, anti-inflammatory response. Then, I thought that macrophages would be in trouble to simultaneously deal with intracellular pathogens such as T. cruzi and remove apoptotic cells generated during infection.
Macrophages, apoptotic lymphocytes and a pathogenic parasite tale
I and my first PhD student tested this hypothesis by studying how parasite-infected macrophages behave after the phagocytosis of apoptotic lymphocytes. Whereas T cells help infected macrophages, which then kill parasites, the interaction with apoptotic cells changes macrophage metabolism and promotes parasite replication within macrophages. We also showed that the injection of apoptotic cells increased parasitemia in T. cruzi-infected mice. For this research published in ‘Nature’1, we received international awards that further promoted our scientific projects and careers.
1Freire-de-Lima, C.G., D.O. Nascimento, M.B.P. Soares, P.T. Bozza, H.C. Castro-Faria-Neto, F.G. de Mello, G.A. DosReis, and M.F. Lopes. 2000. Uptake of apoptotic cells drives the growth of a pathogenic trypanosome in macrophages. Nature 403: 199-203. doi: 10.1038/35003208
My following PhD students investigated which mechanisms were involved in the apoptosis of T lymphocytes in experimental Chagas disease. As a proof of principle, we treated infected mice with apoptosis inhibitors to evaluate the outcomes in T cell-mediated immunity. We found that treatment not only improved the immune responses mediated by T lymphocytes and macrophages but also considerably reduced parasitemia.
We recapitulated the results obtained with apoptosis inhibitors by using T cells from T. cruzi-infected mice cocultured with peritoneal macrophages. The inhibition of T cell apoptosis impacted macrophage phenotypes by shifting M2 macrophages into M1 macrophages, which were more efficient in fighting parasite infection. These findings suggest that T cell help to macrophages is compromised by T cell apoptosis, thereby promoting infection within macrophages.
Current research goals and scientific findings
In 2017, Thaís Rigoni joined this research, which had been carried out by Mariela Piccin and Natália Vellozo. The objective of Rigoni´s thesis was to unveil potential receptors involved in the efferocytosis (detection and phagocytosis) of apoptotic lymphocytes during T. cruzi infection. We finally gained access to the tools to address the role of TAM (Tyro3, Axl, Mer) receptors in T. cruzi infection by employing the Axl and Mer-defective mouse strains.
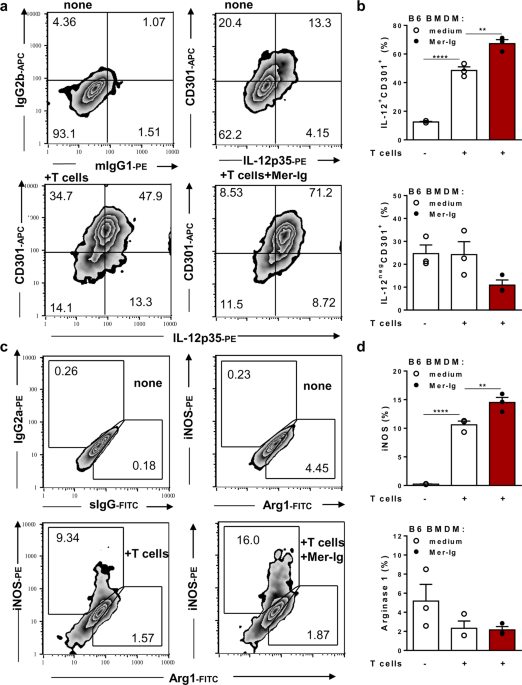
First, we evaluated bone marrow-derived macrophages cultured with T cells from infected mice that provided both helper T cells and proapoptotic T cells. Treatment with a TAM receptor inhibitor not only prevented efferocytosis but also increased hallmarks of M1 activation, such as the expression of induced nitric oxide synthase and nitric oxide production.
We then used synchronized macrophages derived from wild-type (WT), Axl-/- and Mer-/- mice to study the role of each TAM receptor in the efferocytosis of T cells from T. cruzi-infected mice. We found that both Mer (60%) and Axl (30%) accounted for the removal of apoptotic cells by macrophages. Remarkably, Axl defective macrophages expressed the highest M1 responses to helper T cells from infected mice. Moreover, stimulated Axl-/- but not Mer-/- macrophages controlled intracellular infection better than WT macrophages. These findings indicate that Axl suppresses M1 responses and thereby promotes parasite infection.
Axl mediates efferocytosis, promotes T. cruzi infection and heart pathology
Next, we infected WT and Axl-defective mice with T. cruzi to evaluate efferocytosis and immune responses. Peritoneal macrophages from infected Axl-/- mice expressed reduced ability to attach to fluorescent apoptotic cells and to interiorize apoptotic cells. Furthermore, apoptotic T cells accumulated in the spleens of Axl-/- mice compared to their WT counterparts. We then addressed macrophage function during infection and found that macrophages from infected Axl-/- mice showed an increased M1 phenotype and ability to contain parasite infection.
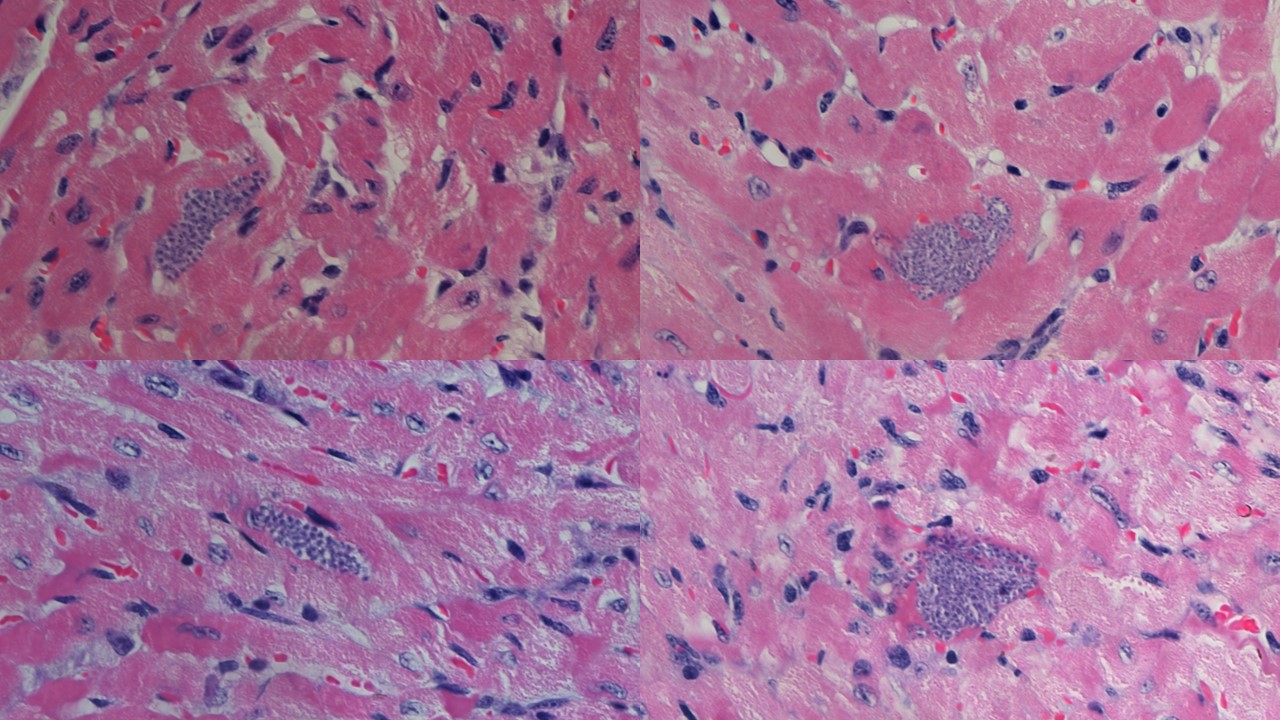
Finally, we investigated how Axl-/- and Mer-/- mice deal with T. cruzi infection and Chagas disease. We showed that parasitemia did not peak in Axl-defective mice in contrast to WT and Mer-/- mice. More interestingly, the hearts of infected Axl-/- mice developed reduced inflammatory responses and fibrosis throughout acute infection. By microscopic inspection of heart sections, we observed parasite nests inside the myocytes (Fig. 1). T. cruzi parasites replicate within these nests (Fig. 1a). We also found a ruptured parasite nest in the heart section from an infected Axl-/- mouse, whereas inflammatory cells approached the nest (Fig. 1b). Furthermore, more macrophages expressed M1 activation in the hearts of infected Axl-/- mice than in those of WT or Mer-/- mice.
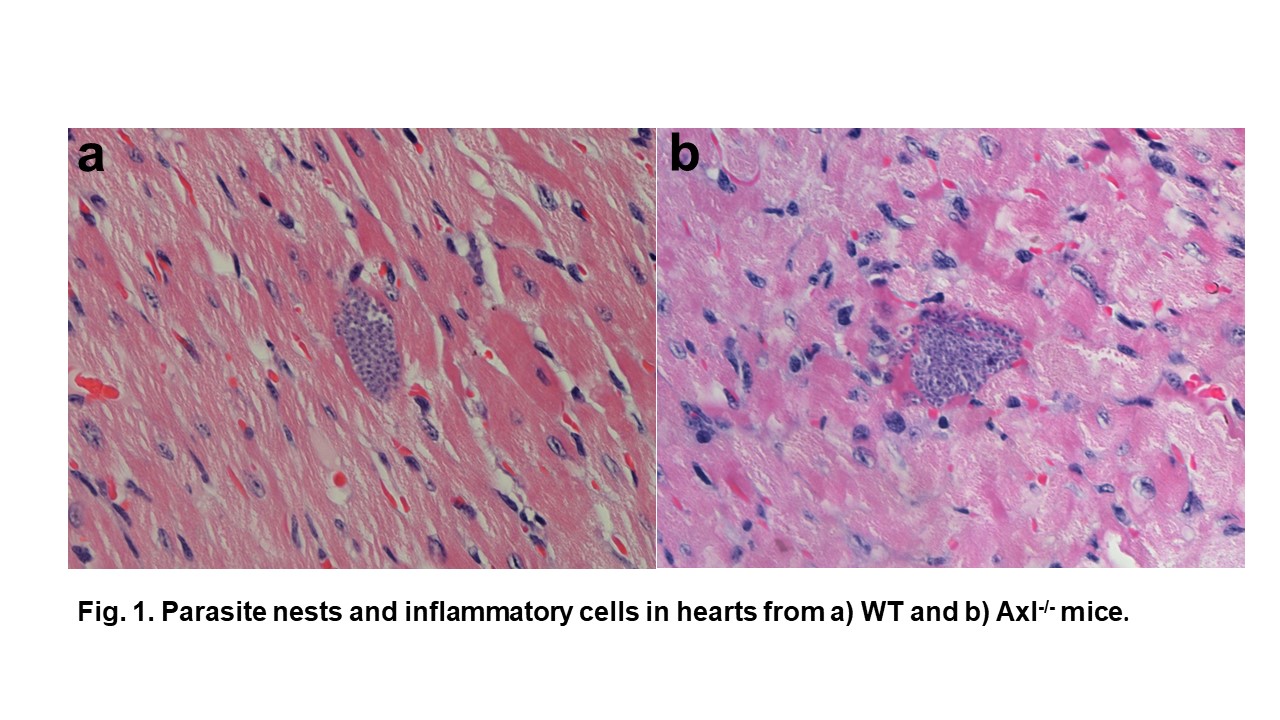
Altogether, these results indicate that Axl disrupts macrophage ability to control parasite infection and thereby contributes to the development of heart pathology in the experimental model of Chagas disease. We expect that the elucidation of the mechanisms involved in pathogenesis will translate into the development of new tools to prevent or treat disease.
This research was conducted by nine women who shared the joy of new discoveries. This work was developed at the Brazilian public university that counted with restricted funding resources in a political scenario which was adverse for science in Brazil.
Marcela F. Lopes
Instituto de Biofísica Carlos Chagas Filho, Universidade Federal do Rio de Janeiro.
Follow the Topic
-
Communications Biology
An open access journal from Nature Portfolio publishing high-quality research, reviews and commentary in all areas of the biological sciences, representing significant advances and bringing new biological insight to a specialized area of research.

Related Collections
With Collections, you can get published faster and increase your visibility.
Artificial Intelligence Methodology in Structural Biology
Publishing Model: Hybrid
Deadline: Nov 30, 2026
Healthy Aging
Publishing Model: Open Access
Deadline: Dec 31, 2026




Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in
Please see https://doi.org/10.3389/fimmu.2023.1244071 for a discussion on the role of M1 and M2 macrophages, T cell apoptosis, and efferocytosis in the pathogenesis of Chagas disease.