Infectious Complications with BCMA-targeting therapies
Published in Biomedical Research and General & Internal Medicine
There are currently a range of treatments for multiple myeloma that target B-cell maturation antigen (BCMA), including chimeric antigen receptor (CAR) T-cells, antibody drug conjugates and bispecific antibodies. Some agents are in ongoing development, whilst many have been approved by the US Food and Drug Administration (FDA). T-cell engaging therapies (CAR T-cells and bispecific antibodies) have a unique safety profile with cytokine release syndrome and neurotoxicity as early common side effects. Additionally, BCMA is expressed on both normal and abnormal plasma cells, and this may contribute toward a generalized state of immune deficiency with the potential for increased risk for infectious complications with these agents. With increasing use of BCMA-targeted therapies in myeloma, and particularly in earlier lines of therapy, the incidence and spectrum of infectious complications observed with these agents warrants close attention. A detailed comparison of infectious complications between BCMA-targeting therapies can serve to optimize current monitoring strategies of immune function, augment infectious prophylaxis and potentially aid in treatment selection. In this single-center, retrospective analysis, we describe the nature, incidence, rate and modifiable risk factors for infectious complications in recipients of BCMA-directed CAR T-cells compared to BCMA-directed bispecific antibodies. A similarly heavily pre-treated cohort of patients who received BCMA-directed antibody drug conjugates were also evaluated. The primary endpoint was the incidence of severe (grade ≥3) infections. Secondary endpoints included infection rate over time, infectious organisms, infectious sites, infection risk factors, and the impact of modifiable risk factors of treatment-emergent hypogammaglobulinemia and neutropenia. All infection-specific events were collected from treatment initiation up to the date of next line of therapy or last follow-up.
256 patients were included in this analysis, of whom 92 received CAR T-cells, 55 bispecific antibodies and 109 antibody drug conjugates. Baseline characteristics of the three treatment arms were overall similar, and the patients were similarly heavily pre-treated with a median of 6-7 prior lines of therapy.
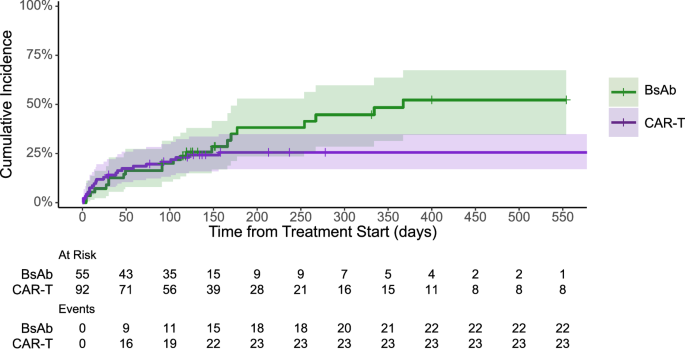
Recipients of BCMA-directed bispecific antibodies had the highest incidence of severe infections (40%), followed by CAR T-cell (26%) and antibody drug conjugates (8%). Whether or not this may relate to chronic stimulation of T-cells and their subsequent exhaustion from continuously dosed bispecific agents remains unclear. The lower infectious risk observed with antibody drug conjugates may also suggest that infection risks could be more attributable to the mechanism of action of specific classes of therapies rather than inherent patient characteristics or the target protein only.
There were no grade 4-5 infections with BCMA CAR T-cells versus 11% with bispecific antibodies. Furthermore, the number of patients who experienced recurrent severe infections was also higher with bispecific antibodies compared to CAR T-cells. In multivariable analysis, there was a significantly lower incidence rate for severe infections with CAR T-cells compared to bispecific antibodies. Together, these findings suggest that recipients of BCMA-directed bispecific antibodies appear to have a higher and more persistent risk of severe infections than their CAR T-cell counterparts.
We also assessed the impact of treatment-emergent hypogammaglobulinemia and neutropenia on infectious events. After initiation of T-cell engaging therapies, hypogammaglobulinemia was commonly seen, and the incidence and incidence rate of severe infections was higher with bispecific antibodies than with CAR T-cells in the presence of hypogammaglobulinemia. Pending confirmation in prospective trials, these results provide a rationale for consideration of intravenous or subcutaneous immunoglobulin in patients receiving BCMA-directed bispecific antibodies.
Regarding the presence of neutropenia, post-therapy neutropenia was more common after CAR T-cell therapy and the presence of neutropenia associated with a significantly higher incidence rate of severe infections with CAR T-cells when compared to periods of non-neutropenia. Therefore, neutropenia appears to play a more significant role in infection severity in CAR T-cell recipients, and this may have implications for growth factor use in this cohort.
In conclusion, we observed an increased susceptibility to severe infections, and more persistent infection risk with BCMA-directed bispecific antibodies compared to CAR T-cells. Our results suggest a higher infection risk with bispecific antibodies during periods of hypogammaglobulinemia, and during periods of neutropenia with CAR T-cells, and these findings may have implications for distinct supportive care strategies for these agents. Finally, future clinical trials of BCMA directed T-cell engaging therapies should report infections and the impact of hypogammaglobulinemia, neutropenia and immune function in finer detail to better understand infectious risks and thereby develop appropriate mitigation strategies.
Follow the Topic
-
Blood Cancer Journal
This journal seeks to publish articles of the highest quality related to hematologic malignancies and related disorders.

Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in