Inflammasome activation under high cholesterol load triggers a protective microglial phenotype while promoting neuronal pyroptosis
Published in Neuroscience
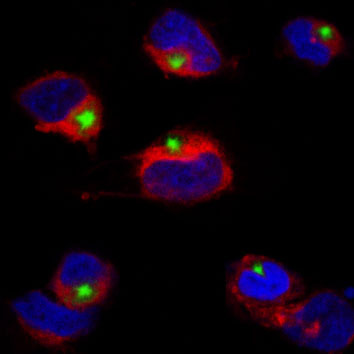

Neuroinflammation and progressive neuronal death are two critical hallmarks of Alzheimer’s disease (AD). Still, the particular mechanisms underlying these alterations have not been fully characterized. To date, researchers have mainly focused on deciphering the role of pro-inflammatory microglia; however, mounting evidence points to a more complex scenario with intricate interconnections between dysfunctional glial cells and dying neurons. In our study, recently published in Transl Neurodegener, we pursued regulatory factors that may govern these interrelationships and thus be ultimately responsible for chronic inflammation.
Key players of the innate immune response are inflammasomes, molecular assemblies activated in response to damage- (DAMPs) or pathogen- (PAMPs) associated molecular patterns, which lead to caspase-1 activation, the subsequent cleavage maturation of pro-inflammatory interleukins IL-1β and -18, and under some circumstances the activation of gasdermin D (GSDMD), executor of pyroptosis, an inflammatory-type of cell death. Inflammasome activation has been observed in experimental models and individuals with AD. Nonetheless, despite the link with the neurodegenerative process, how the pathway is regulated is largely unknown.
It had been described that mitochondrial oxidative stress can be sensed by the pattern recognition receptor NLRP3, triggering the inflammasome assembly. Remarkably, mitochondrial dysfunction and oxidative stress are both early events in the progression of AD, which, as described in previous studies by the group, become aggravated when intracellular cholesterol rises and results in depleted mitochondrial glutathione (GSH), the main antioxidant defense of mitochondria. Taking into consideration these data, we decided to evaluate whether the cholesterol and amyloid beta (Aβ)-induced oxidative stress can regulate inflammasome activation in microglia and neurons. We also studied the functional crosstalk between both cell types when inflammasome is engaged, setting new paradigms and potential therapeutic targets for AD.
In microglia, after inflammasome engagement, we found that cholesterol enrichment promoted the release of IL-1β encapsulated in extracellular vesicles. The functional significance of this change is uncertain but may likely influence cell-cell communication. In addition, the increased cholesterol load also favored the conversion of activated microglia to a high phagocytic phenotype, with increased expression levels of Trem2 and Clec7, thus resembling disease-associated microglia (DAM), a subset of resident macrophages recently identified and related to neurodegeneration. Conditioned media from these cells displayed high levels of neurotrophins and, when used in neuronal cells, exerted a protective effect against Aβ-induced cytotoxicity, reinforcing the link between high cholesterol levels and a neuroprotective microglial behavior.
In parallel, inflammasome signaling was analyzed in cholesterol-enriched SH-SY5Y neuroblastoma cells exposed to bacterial endotoxins and Aβ. In both cases, increased intracellular cholesterol levels stimulated inflammasome assembly and caspase-1 activation. Unlike microglia, however, the enhanced inflammasome induction in neurons was accompanied by overt GASDMD-mediated pyroptosis.
Having previous works shown that oxidative stress contributes to inflammasome activation, we wanted to investigate whether mitochondrial GSH depletion in cholesterol-enriched cells was responsible for the enhanced induction of the pathway. To get to that extent, we treated the cells with GSH ethyl ester (GSHee), a membrane-permeable form of GSH that significantly reduced the oxidative stress evoked by Aβ. The antioxidant effect of GSHee prevented inflammasome assembly and the translocation of GSDMD to the plasma membrane, which resulted in reduced cell death.
Finally, we evaluated whether cholesterol-promoted pyroptosis in neuronal cells after Aβ exposure can affect microglia function. We incubated microglia with conditioned media of Aβ-treated neuronal cells with and without cholesterol enrichment and analyzed their phagocytic capacity. We found that media from pyroptotic cells severely affected the Aβ uptake of microglia. Remarkably, the inhibitory effect on phagocytosis was not appreciable when we used conditioned media from cells pre-treated with GSHee, which prevented neuronal pyroptosis.
Our findings indicate that the microglia-neuron communication can ultimately modify microglia behavior towards a less protective phenotype, which is in line with growing evidence suggesting that the loss of microglial protection (dystrophic microglia), as opposed to reactive microglial (pro-inflammatory), is the trigger for the cascade of events that leads to sustained neuroinflammation in AD.
We propose a model in which cholesterol levels act as a checkpoint of the immune response in AD, regulating the signaling pathways driven by inflammasome differentially in microglia and neurons, thus favoring a neuroprotective phenotype in microglia ultimately challenged by a pro-inflammatory neuronal death when intracellular cholesterol content rise and mitochondrial GSH is depleted.

Follow the Topic
-
Translational Neurodegeneration
An open access, peer-reviewed journal that covers research, therapeutics and education for all aspects of neurodegenerative diseases.





Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in