Machine learning insights into predicting biogas separation in metal-organic frameworks
Published in Chemistry, Materials, and Statistics
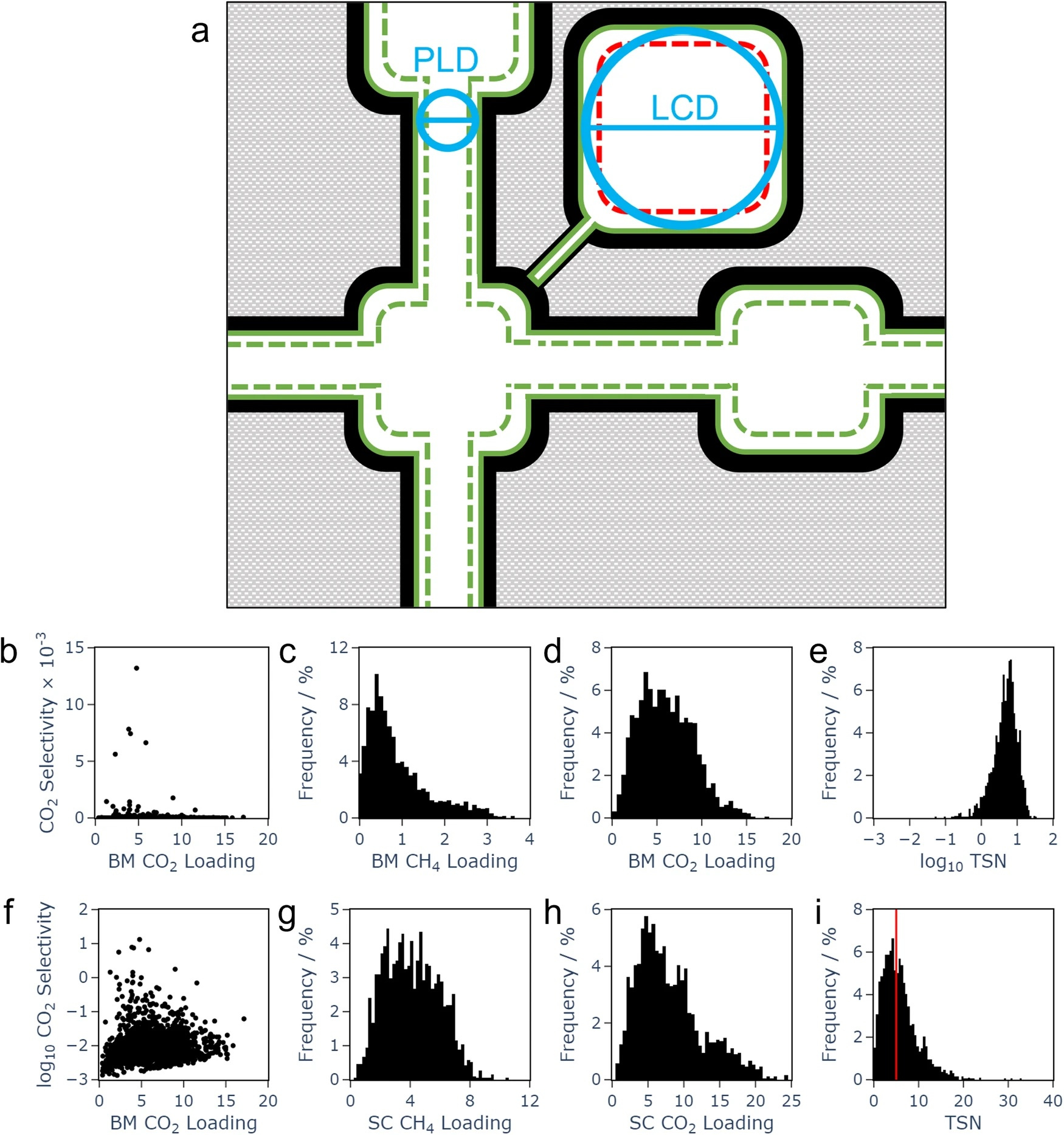
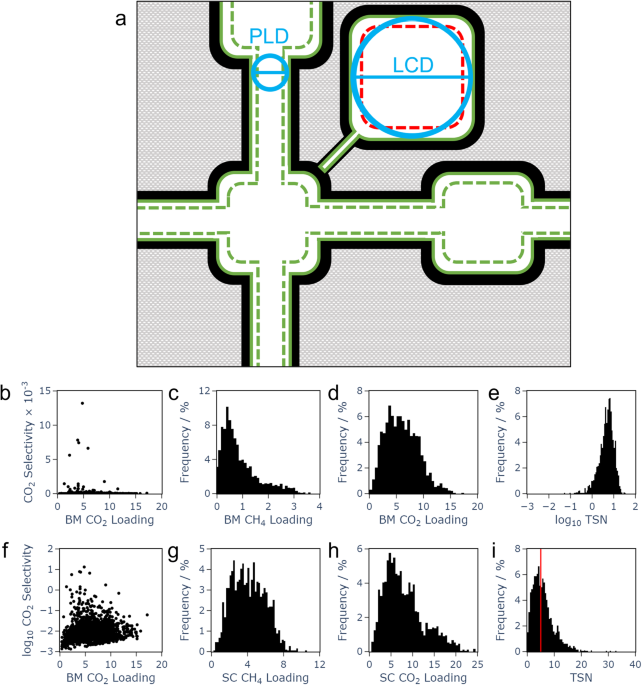
With the modern world increasingly impacted by the effects of climate change, it is imperative to pursue all available avenues for increasing sustainability across all sectors. One of these avenues is the use of biogas as a fuel alternative. It burns more cleanly than fossil fuels and can be obtained from renewable sources, in particular existing waste stocks from agricultural and industrial processes. However, a challenge when it comes to achieving more widespread use of biogas as fuel is obtaining sufficiently pure gas streams. The cleanest and most efficient biofuel is composed of pure methane, CH4, but biogas sources contain contaminants including CO2. Efficient, effective, and sustainable methods to separate CO2/CH4 streams and upgrade the biogas are needed. One possible method is separation by an appropriate porous material. Adsorption, the gathering of molecules on the material’s surface, is key here. A well-selected material can adsorb CO2 while leaving CH4 largely free to pass, and the process is enhanced by specific chemical interactions. In this work, we leveraged computational chemistry and machine learning methods to search material databases for promising materials based on predicted properties.
The materials we focused on are metal organic frameworks (MOFs). These crystalline solids have specific structures, with fragments containing metal atoms bonding to those containing carbon. By altering the chemical makeup of the fragments, their bonding geometry, or a combination of the two, it is theoretically possible to access hundreds of thousands of MOFs with different properties. Certain MOFs perform separations very well for specific targeted gas mixtures with a separation process which depends on tailored chemical interactions.
Finding a MOF with excellent performance among the many possibilities is a formidable task; experimentally creating and testing even a small fraction of the potential structures is not possible. However, several databases exist populated with the chemical structures of large numbers of MOFs. These databases are a valuable tool. Through them, the field lends itself to studies involving fast property prediction for thousands of accessible structures, allowing refinement and material selection. Computational statistical thermodynamics methods using a sufficiently accurate underlying model can be used to make reasonable predictions, but they are limited by their speed. Machine learning models can be trained to reproduce their results at greater speed, expediting database searching and accelerating the development of new materials for biogas upgrading.
Useful as they are, some of the information contained in these databases is flawed: some of the structures defy chemical rules and do not represent realistic materials. The scale of this issue is surprising given MOF databases are ultimately based on experimentally characterised structures. However, the problems exist in all major MOF databases, affecting a large fraction of entries, often over 50%. They come about as a result of dealing with data on a large scale using automatic processing methods which miss chemical nuance. Where unreasonable chemical structures are present in materials searches, they can lead to misleading results, particularly if they are used to train machine learning models. We curated the databases that we used in our work, applying several checks to the chemical structures and removing unreasonable data.
Once suitable databases were selected and unreasonable structures removed, we used computational chemistry methods based on statistical thermodynamics to predict the ability of the MOFs to separate CO2 and CH4. We looked for MOFs expected to adsorb a high total quantity of CO2 while keeping CH4 adsorption low, and we picked out a handful of MOFs based on this. We also identified geometrical properties displayed by promising MOFs to help guide future searches.
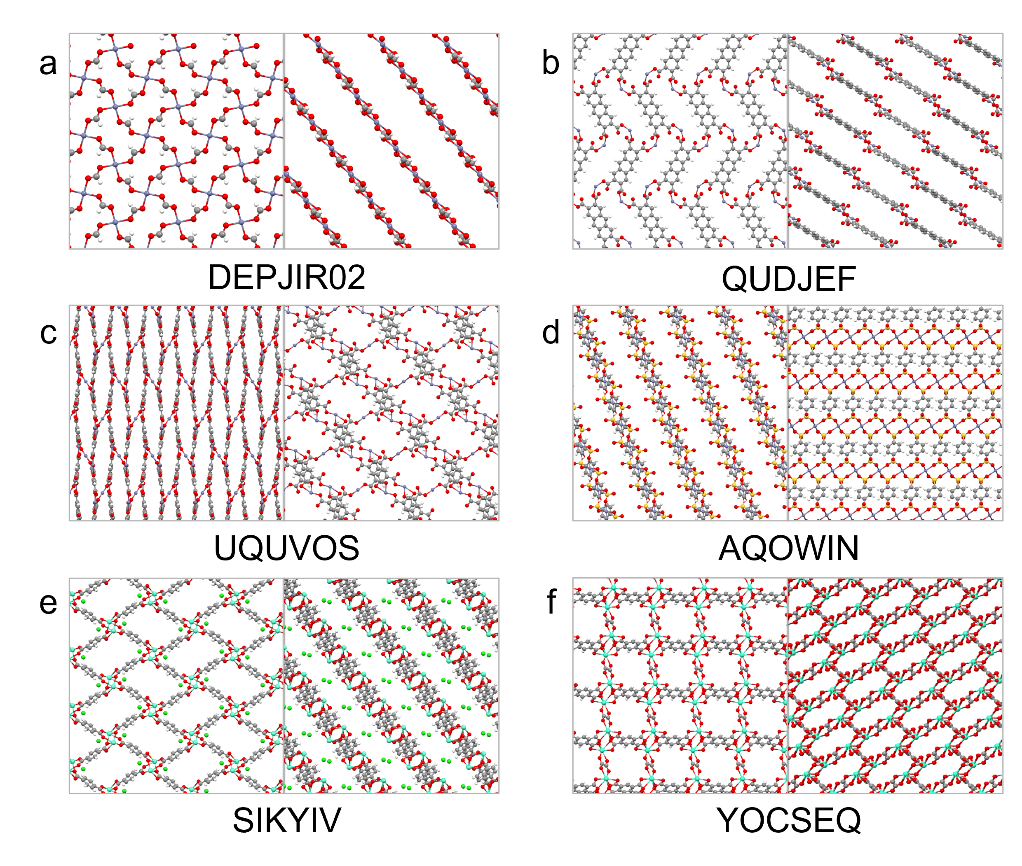
Figure 1: Chemical structures of six promising MOFs selected by our computational chemistry search.
Calculations of this kind, while faster than experiments, can take hours or even days, leading to a sizeable total simulation time. Therefore, we used the results from this property search – chemical structures of thousands of MOFs and predictions about their separation ability – as input data to train machine learning models to speed up future similar property searches. These mathematical models find relationships between readily available data about a material and other less accessible data. They can then be used for new materials to almost instantly make predictions about the less accessible data. In this case, the models make predictions about separation ability based on chemical structure and other readily available information. The models we trained were of two main types, regression and classification. The first make numerical predictions meaning that, if of high enough quality, they can be highly nuanced and specific. The second were trained to identify each MOF as either high-performing or low-performing, and may be used in the early stages of a property search to eliminate low-performing MOFs before any more laborious calculations are needed, focusing efforts on the best materials. We trained a variety of different kinds of both regression and classification models for comparison.
The models performed well on the training and validation set from our computational chemistry search. We selected the most accurate of them and tested them on a set of unseen MOFs from a different database. We still saw strong performance, although, as expected, accuracy was reduced compared to the training performance. In doing these tests, we selected MOFs from the test set identified by the machine learning models as those most likely to display strong performance.
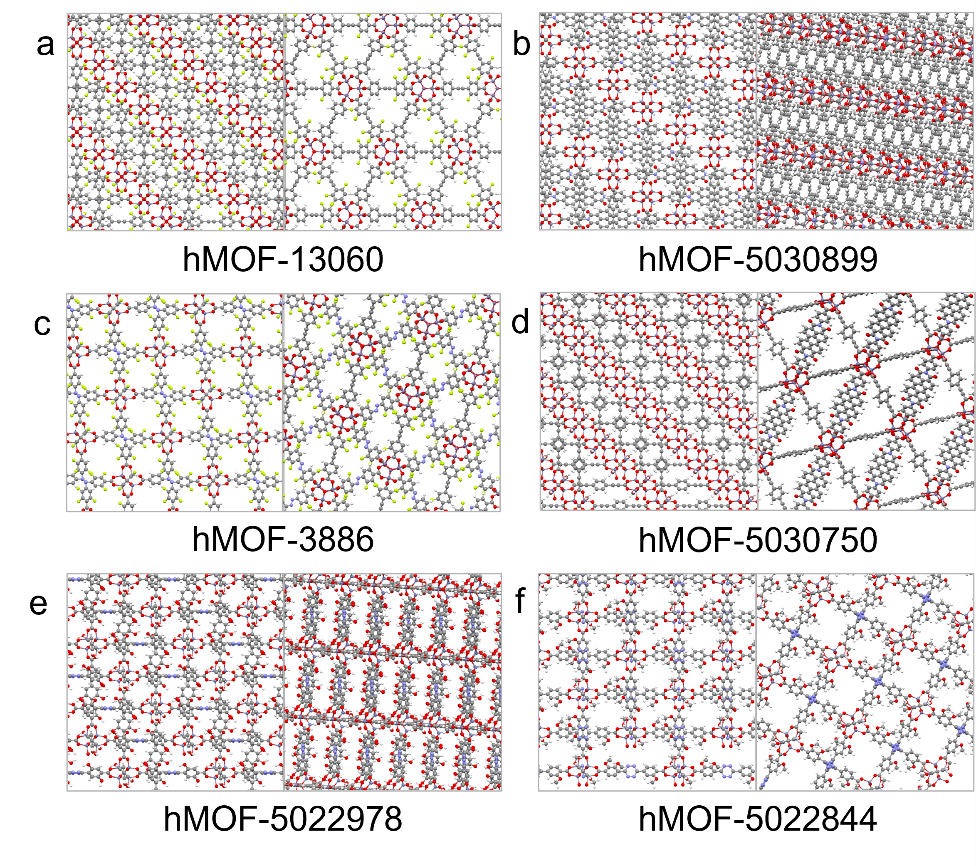
Figure 2: Chemical structures of six promising MOFs selected by our machine learning search.
In order to best understand the models and pave the way for their future improvement, we extensively analysed their accuracy in the context of the data. We compared the models to each other, identified the kinds of MOFs for which predictions were most accurate, and proposed ways that accuracy could be expanded in future work. In the interests of open access and reproducibility, all relevant scripts and datasets from this work are available and downloadable from a GitHub repository.
Overall, in this work we have identified promising MOFs which may promote the sustainable purification of biogas mixtures. We have also made available fast and efficient methods which may be used to expedite similar structure searches in future. Therefore, this work can help to unlock the potential of biogas fuel in promoting sustainability and waste reduction across several sectors.
Follow the Topic
-
Communications Chemistry
An open access journal from Nature Portfolio publishing high-quality research, reviews and commentary in all areas of the chemical sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Chemical modification of proteins
Publishing Model: Open Access
Deadline: Sep 30, 2026
Sustainable waste management through polymer upcycling
Publishing Model: Open Access
Deadline: Aug 31, 2026


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in