Matched patient biopsies offer insight into resistance mechanisms in prostate cancer
Published in Cancer

Background
Development of resistance to targeted therapies is nearly universal in cancer treatment. However, there is an incomplete understanding of resistance mechanisms for most cancer therapies due to the paucity of data utilizing matched biopsies from patients treated with these agents. For patients with prostate cancer, androgen deprivation therapy with drugs like the androgen receptor (AR) inhibitor enzalutamide (enza) is the principal treatment approach, but much remains unknown about how this drug stops working.
Prostate cancer is the second leading cause of cancer related death in men in the United States, with 34,500 deaths predicted for this year alone1. Nearly all prostate cancers are adenocarcinomas at diagnosis and driven by the AR2. Androgen deprivation therapy (ADT) has been the principal treatment metastatic prostate cancer for over 80 years. Eventually resistance to ADT develops, though the majority of metastatic castration resistant prostate cancers (CRPC) remain androgen-dependent3. This led to the development of more potent AR inhibitors such as enza. Most patients benefit from enza4,5, but disease progression is inevitable. Previous studies implicated several resistance mechanisms, including continued AR-dependence on one end of the spectrum vs. reduced AR-dependence and lineage plasticity, or differentiation change, on the other end6-9.
Lineage plasticity is defined as a biologic process that occurs during normal development and later as a mechanism that promotes survival when cells are under stress. Plasticity may manifest as reversible or irreversible changes in cellular “identity,” whereby cells take on an alternative phenotypic or epigenetic state10. Lineage plasticity in prostate cancer is characterized by loss of canonical AR signaling. However, lineage plasticity is likely a continuum, ranging from AR activity-low tumors that remain adenocarcinomas to those that lose AR expression and express a neuroendocrine or neuronal program known as neuroendocrine prostate cancer (NEPC), and finally those that lose AR expression but do not express a neuroendocrine or neuronal program (i.e., double negative prostate cancer (DNPC)11,12.
Transcriptional assessments of AR activity and lineage
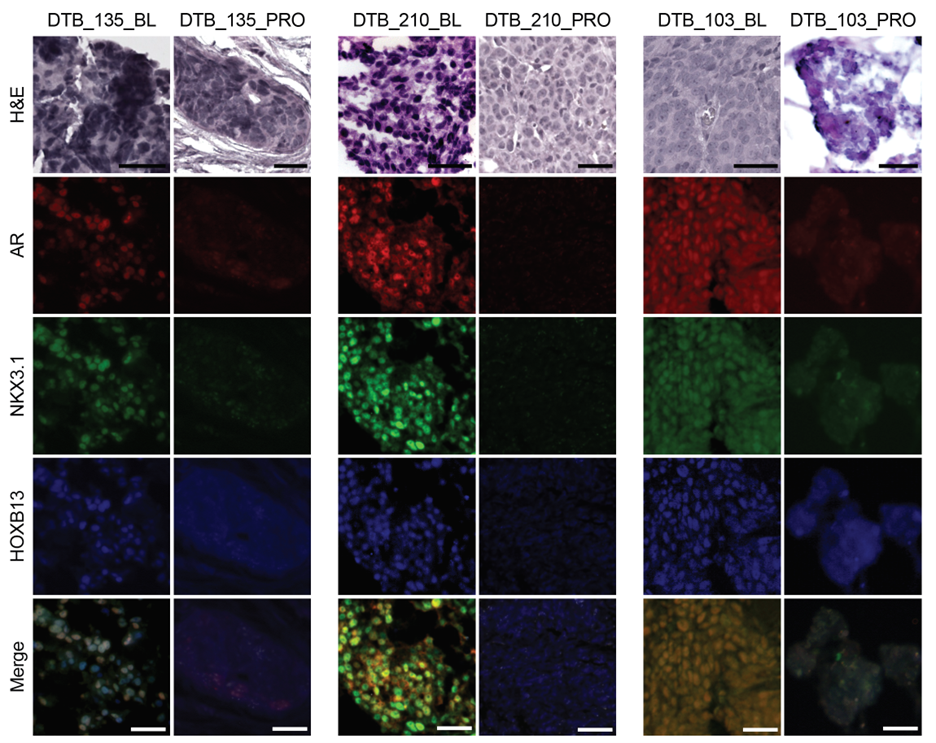
To clarify emergent enza resistance mechanisms in patients with CRPC, we initiated a longitudinal study with metastatic biopsies prior to enza treatment and at the time of progression (Figure 1)13. Twenty-one patients had matched biopsies performed that underwent laser capture microdissection to enrich for tumor cells prior to RNA-sequencing. All samples were AR-dependent adenocarcinomas at baseline. However, at the time of progression, three tumors were classified as DNPC based on multiple transcriptional measures and immunofluorescence.

Figure 1. Study Schema
Development of a lineage plasticity risk signature score
We hypothesized that the baseline samples from the three patients whose tumors eventually converted to DNPC would harbor a unique transcriptional profile compared to the other baseline samples that remained AR-driven at progression. Indeed, we identified a gene signature that was highly expressed in these three baseline tumors vs. the other 18 tumors that did not undergo lineage plasticity. A cutoff was established based on the lowest score of the three converters. Suitable validation cohorts with matched biopsies did not exist, but we further hypothesized that high expression of this signature would be linked not only to lineage plasticity risk but also poor clinical outcomes. Therefore, we first applied our lineage plasticity risk signature to two independent cohorts of patients with CRPC for whom time on AR inhibitor treatment was available. High lineage plasticity scores using our pre-defined cutoff was strongly associated with poor survival (Figure 2).
We previously determined the effect of surgical castration on risk of lineage plasticity in a collection of human patient-derived xenografts (PDX) tumors. Only one tumor, termed LTL331 undergoes castration-induced lineage plasticity14. Importantly, our lineage plasticity risk signature was highly expressed in LTL331 vs. the other PDXS (Figure 3). These results strongly suggest that the signature we identified is linked to poor patient outcome and risk of lineage plasticity upon AR inhibition.


castration-naïve adenocarcinoma PDX models described by Lin, et al14.
Only LTL331 undergoes castration-induced lineage plasticity
Transcriptional changes and DNA profiling in tumors undergoing lineage plasticity
The three samples that underwent lineage plasticity showed loss of AR expression and AR activity without upregulation of classical neuroendocrine markers—most consistent with DNPC. Pathway analysis showed enrichment in several inflammatory pathways including: allograft rejection, interferon gamma and alpha response, and IL6/JAK/STAT signaling. These pathways have been identified in tumor stem cells15,16, and we postulate that the progression tumors that underwent lineage plasticity have lost a luminal differentiation program and become more stem-like. Finally, DNA sequencing studies showed conservation of mutations between baseline and progression samples from converters, strongly suggesting that outgrowths of genetically distinct clones did not account for lineage plasticity and raising the possibility of epigenetic changes.
Resistance mechanisms in the wider cohort
What about the eighteen patients whose progression tumors did not undergo lineage plasticity? Importantly, all the patients in our cohort, including these 18 patients, were still taking enza at the time of their progression biopsy. We found that almost half of these patients had similar AR activity between their baseline and progression samples, suggesting that maintenance of AR function despite enza treatment explained disease progression. Though there was variability in AR function in the remaining progression samples, we observed upregulation of many of the same stemness pathways found to be upregulated at progression in the tumors that converted to DNPC. This suggests that this subset of patients had increased stemness in their tumors at progression without fully losing AR expression and AR signaling.
Future directions
A natural next step from this work is a more careful dissection of mechanisms that contribute to lineage plasticity risk, including chromatin or microenvironmental contributors. Furthermore, it will be important to attempt to validate the lineage plasticity risk signature we identified in additional independent cohorts with matched biopsies. Once biomarkers of lineage plasticity risk are identified, prospective clinical trials testing drugs that target pathways highly activated in these tumors may be warranted, including studies with agents such as BET bromodomain inhibitors17 or JAK/STAT inhibitors18.
The treatment paradigm in metastatic prostate cancer has changed since our study was completed. Patients now often receive enza and other AR signaling inhibitors upfront along with ADT, instead of at the time of progression on ADT. Our work did suggest that the lineage plasticity risk signature we identified is extremely uncommon in hormone therapy-naive primary prostate cancer tumors from The Cancer Genome Atlas cohort. Therefore, it may be necessary to determine whether other markers exist to predict risk of lineage plasticity from the get-go, work we are currently undertaking.
References
- Siegel, R.L., Miller, K.D., Fuchs, H.E. & Jemal, A. Cancer statistics, 2022. CA Cancer J Clin 72, 7-33 (2022).
- Humphrey, P.A. Histopathology of Prostate Cancer. Cold Spring Harb Perspect Med 7(2017).
- Montgomery, R.B., et al. Maintenance of intratumoral androgens in metastatic prostate cancer: a mechanism for castration-resistant tumor growth. Cancer Res 68, 4447-4454 (2008).
- Beer, T.M., et al. Enzalutamide in metastatic prostate cancer before chemotherapy. N Engl J Med 371, 424-433 (2014).
- Scher, H.I., et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N Engl J Med 367, 1187-1197 (2012).
- Abida, W., et al. Genomic correlates of clinical outcome in advanced prostate cancer. Proc Natl Acad Sci U S A 116, 11428-11436 (2019).
- Aggarwal, R., et al. Clinical and Genomic Characterization of Treatment-Emergent Small-Cell Neuroendocrine Prostate Cancer: A Multi-institutional Prospective Study. J Clin Oncol 36, 2492-2503 (2018).
- Alumkal, J.J., et al. Transcriptional profiling identifies an androgen receptor activity-low, stemness program associated with enzalutamide resistance. Proc Natl Acad Sci U S A 117, 12315-12323 (2020).
- Annala, M., et al. Circulating Tumor DNA Genomics Correlate with Resistance to Abiraterone and Enzalutamide in Prostate Cancer. Cancer Discov 8, 444-457 (2018).
- Beltran, H., et al. The Role of Lineage Plasticity in Prostate Cancer Therapy Resistance. Clin Cancer Res 25, 6916-6924 (2019).
- Bluemn, E.G., et al. Androgen Receptor Pathway-Independent Prostate Cancer Is Sustained through FGF Signaling. Cancer Cell 32, 474-489 e476 (2017).
- Labrecque, M.P., et al. Molecular profiling stratifies diverse phenotypes of treatment-refractory metastatic castration-resistant prostate cancer. J Clin Invest 129, 4492-4505 (2019).
- Westbrook, T., et al. Transcriptional Profiling of Matched Patient Biopsies Clarifies Molecular Determinants of Enzalutamide-Induced Lineage Plasticity. Nat Commun In print(2022).
- Lin, D., et al. High fidelity patient-derived xenografts for accelerating prostate cancer discovery and drug development. Cancer Res 74, 1272-1283 (2014).
- Lo, U.G., et al. IFNgamma-Induced IFIT5 Promotes Epithelial-to-Mesenchymal Transition in Prostate Cancer via miRNA Processing. Cancer Res 79, 1098-1112 (2019).
- Yamashina, T., et al. Cancer stem-like cells derived from chemoresistant tumors have a unique capacity to prime tumorigenic myeloid cells. Cancer Res 74, 2698-2709 (2014).
- Aggarwal, R.R., et al. A Phase Ib/IIa Study of the Pan-BET Inhibitor ZEN-3694 in Combination with Enzalutamide in Patients with Metastatic Castration-resistant Prostate Cancer. Clin Cancer Res 26, 5338-5347 (2020).
- Chan, J.M., et al. Lineage plasticity in prostate cancer depends on JAK/STAT inflammatory signaling. Science, eabn0478 (2022).
Follow the Topic
-
Nature Communications
An open access, multidisciplinary journal dedicated to publishing high-quality research in all areas of the biological, health, physical, chemical and Earth sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Women's Health
Publishing Model: Hybrid
Deadline: Ongoing
Biosensing
Publishing Model: Hybrid
Deadline: Sep 30, 2026




Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in