Molecules "imagined" using advanced artificial intelligence show promising results in pre-clinical experiments
Published in Bioengineering & Biotechnology

For readers unfamiliar with the drug discovery and development process, it is worthwhile to point out that the pharmaceutical industry is one of the most inefficient and risky industries on the planet. The efficiency of the industry has been on the decline since the 1950s. It costs over $2.6 Billion to bring the New Molecular Entity (NME) to the market. And despite the many advances disrupting other industries including personal computing, the Internet, and genome sequencing, the cost to develop a drug is steadily increasing. This is one of the reasons why most industry experts are skeptical about the promises of deep learning. Pharmaceutical veterans have seen promising technological breakthroughs that have not significantly improved R&D and Therefore, they prefer to incrementally develop internal capabilities across the entire spectrum of the drug discovery process instead of making big bets on specific enabling technologies. Readers who would like to get a quick introduction to the drug discovery process should refer to the paper by Steven Paul and his colleagues from Eli Lilly titled “How to improve R&D productivity: the pharmaceutical industry’s grand challenge” published in 2010. Figure 1 below shows the failure rates, time and costs of designing the molecules for a given target.
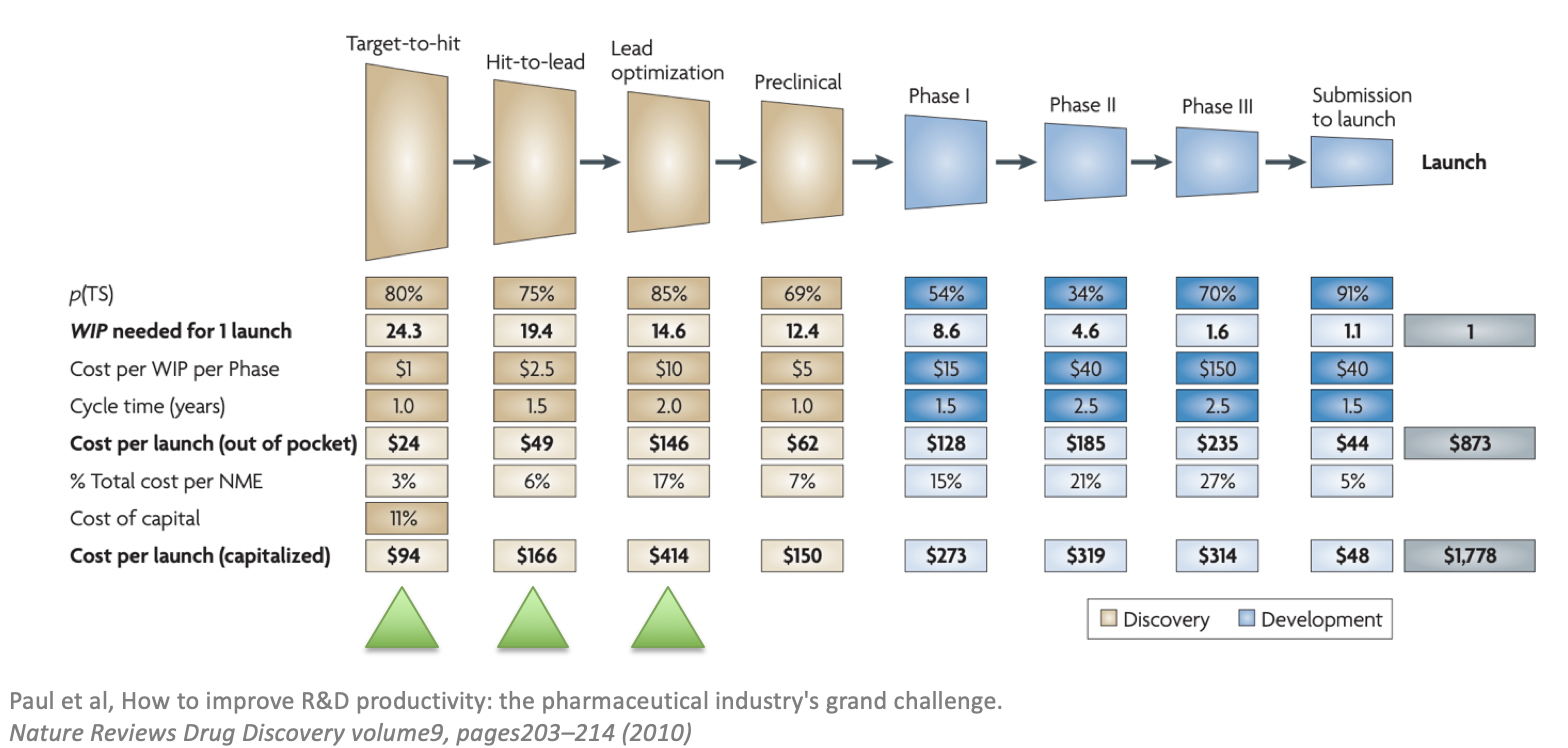
Figure 1: Overview of averaged costs associated with the main stages of drug discovery and development. Green triangles are the phases GENTRL may help accelerate.
The concept of Generative Adversarial Networks (GANs) is relatively new and is often referred to as the “AI imagination”, "creative AI", or "AI curiosity". While some of the ideas may be traced to 1990s, the first paper on "Generative Adversarial Nets" was published in 2014 by Ian Goodfellow, now referred to as the "Father of GANs". Conceptually it is a competition between two deep neural networks, where one, the generator, is generating novel content with the desired set of criteria and another, the discriminator, is testing whether the output of the generator is true or false. In 2016 multiple groups using GANs started producing new photorealistic images from natural language. For example, one could give a description: “this small bird has a pink breast and crown, and black primaries and secondaries” and the GAN would generate or “imagine” a large number of images of birds with said properties.
Starting in 2015, our group started working on an application to incorporate GANs and the generation of novel chemical structures or molecules. When generating pictures, GANs require high-dimensional data and large, well-annotated training sets. Molecules can be represented in the low-dimensional format like binary fingerprints, SMILES strings, graphs and other light representations that can be used to synthesize the resulting molecules.
In our paper titled “The cornucopia of meaningful leads: Applying deep adversarial autoencoders for new molecule development in oncology” which was submitted in June 2016, we described the concept of using the Adversarial Autoencoder (AAE) for the generation of novel molecules. A similar idea in the paper titled “Automatic chemical design using a data-driven continuous representation of molecules“ was put on ArXiv 2016 by Alan Aspuru-Guzik’s team, which also pioneered several areas in quantum chemistry and generative materials science. At that time my team was very disappointed since our paper was still in review and it took two months to find the reviewers in a reasonably fast journal. But then we realized that this is a marathon and started collaborating to build a community around generative chemistry.
Since then we have published a very large number of generative approaches and also started combining them with deep reinforcement learning, a form of AI learning strategy that was used in AlphaGo to defeat a human Go champion.

Figure 2: (Top) Timeline summarizing the key advances towards the development of machine and deep learning. (Bottom) Timeline of the release of the successive GAN-based models. Insilico Medicine was among the first company to publish the proof of concept of such models for molecular generation and the company has published several models for that purpose during the last five years. (Source: Insilico Medicine: a brief history of deep generative models for de-novo molecular design)
From 2016 to 2018 we presented at over 100 academic and industry conferences. At first, many computational chemists and medicinal chemists in the pharmaceutical industry were skeptical about this novel technology. The molecules that we generated using the generative models were not diverse enough or easy to synthesize and the targets were too easy with the available structures and hits. Since it’s necessary o validate new technologies, we launched a “Tinder for molecules” called Chemistry.AI where we showed how generated molecules and molecules from high-value libraries can improve our generative pipelines using human intuition. Several medicinal chemists who were originally fierce skeptics of GANs and allergic to the word "AI" joined the effort after reviewing the generated molecules in details. For some of them getting the GANs to generate meaningful and modern chemistry and critiquing the output became an obsession. We found it extremely valuable to have active and retired professional medicinal chemists with decades of experience and "mixed martial arts (MMA) careers" in biology and chemistry to work with the deep learning teams. Senior medicinal chemists retiring from the top pharmaceutical companies can always find a consulting opportunity with our team after passing the exam. We realized that there are very few of them and their knowledge is essential for design and validation of the generative chemistry pipelines.
When using GANs to generate images, text or music, it is possible to validate in real-time using the author's sensory organs. In chemistry, one needs to synthesize the molecules, perform in vitro enzymatic assays, metabolic stability assays, disease-relevant assays and then start animal validation and human testing. The feedback loop for confirming whether the generation conditions were met is a long and expensive process; typically there’s a limited number of attempts to get it right because it costs hundreds of thousands of dollars to validate. It can be compared to launching a satellite into orbit for the first time. In 2018 we launched our “Sputnik” with the publication of the first JAK3 inhibitor generated using the Entangled Conditional Autoencoder (ECAAE) with experimental validation. At that time we could already achieve reasonable hit rates for GPCRs and other target classes with our internal generative pipelines that also generate crystal structures.
Traditionally we do most of our synthesis and experiments by working with a company which acts as an open research platform with tens of thousands of chemists that can synthesize pretty much every molecule. We see them as the "Ferrari of synthetic chemistry" and they employ some of the smartest synthetic and medicinal chemists we got to work with to date. After the publication of JAK3 inhibitor, their R&D management team suggested that we perform an “AlphaGo experiment” by timing how long it would take for a generative system to design molecules for an arbitrary target that can be tested in vitro and subsequently rapidly synthesized. We partnered with Alan Aspuru-Guzik, one of the luminaries in the field, and agreed that we would target a kinase using the Generative Tensorial Reinforcement Learning (GENTRL) system that was developed in 2017. We made the code which is internally considered to be a legacy system publicly available for everyone to reproduce and experiment in this direction. Drawing the analogy with Sputnik, it makes sense to publish the blueprints for the original rocket when you already developed the space station. And the basic blueprints are shown in Figure 3 below.
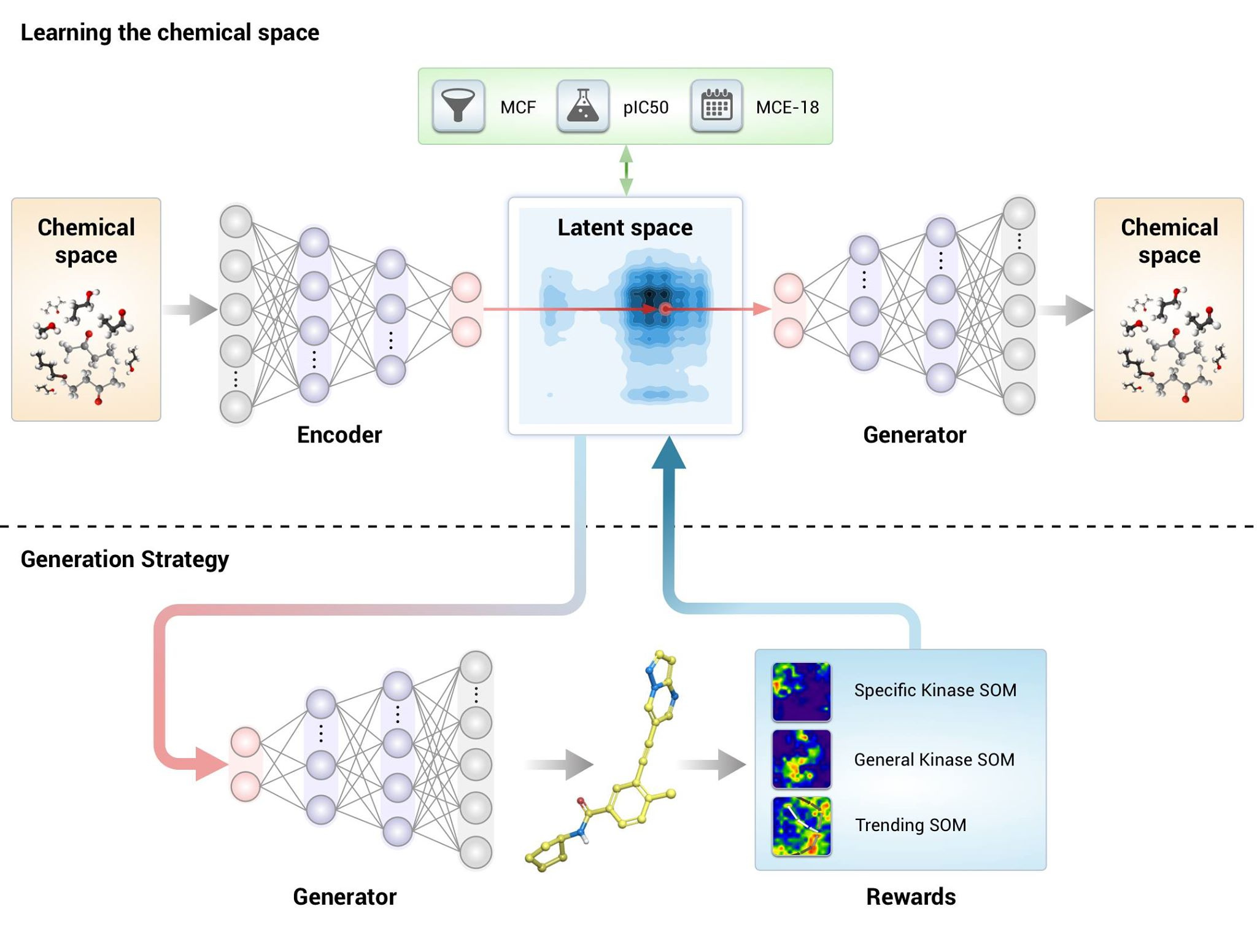
Figure 3: The basic graphical representation of GENTRL approach. It generates the molecules with specific conditions and learns to generate molecules with the specific objectives.
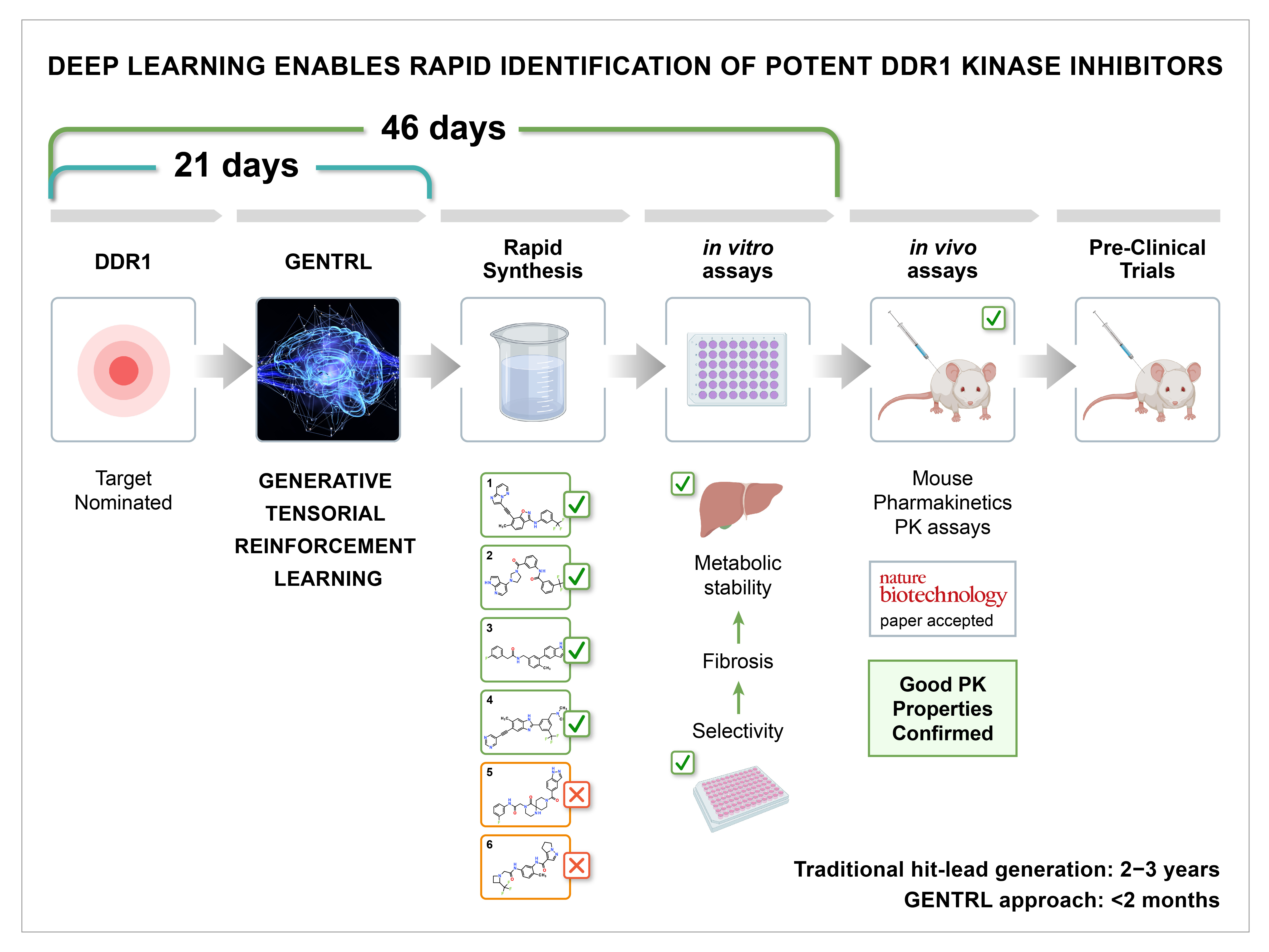
Fast-forward to July 2018, the race was on. DDR1 target was nominated. Since we were unfamiliar with this target and the GENTRL pipeline wasn’t automated, we spent a week gathering data for training. We then spent 12 days training GENTRL and generating the molecules. After 19 days the generated structures were prioritized using the medicinal chemistry filters and 6 molecules were sent for synthesis and further testing. The AI work stopped and the computational scientists held their breath.
Although it was hot in August and many people in South East Asia took a week off, our determined chemistry partners continued with the race and quickly synthesized the molecules and sent them for testing at an external laboratory. It was important to keep the experiment clean for internal validation. By day 46 we received promising results; 4 of the 6 molecules provided were hits and in the nanomolar range. Two of them were single-digit or low double-digit nanomolar inhibitors. These two had good metabolic stability in several assays and worked well in the in- vitro fibrosis assays. That’s when the celebration started!.
The reviews were mixed when we submitted our findings on November 1st. Experts in AI and generative chemistry recommended immediate publication while the experts in drug development were skeptical doubting the metabolic stability of the most potent hits. They requested reproducibility of some of our experiments. The experiments were repeated and confirmed the initial results which were a success.
Even though it is rare to have molecules with adequate metabolic stability in mice at these early stages, we decided to test the pharmacokinetics. Again, the team held their breath. But the results came back with favorable pharmacokinetics.
In parallel, to make this program commercially-viable, we synthesized several DDR1 and DDR2 inhibitors with different properties and selectivity profiles, performed the same set of experiments and started validation in several forms of cancer. We hope to be able to publish these results in the future.
In brief, this study represents a valuable experimental validation showing the promising generative reinforcement learning generator technology used to design the novel molecules that were synthesized and tested in multiple experiments including animals.
This study and GENTRL approach have many limitations. Firstly, the molecules we tested were generated with synthetic accessibility in mind and there are better and more specific molecules out there. Secondly, the DDR1 kinase target is not new and there is plenty of data to train on. It gets trickier when it comes to difficult targets like ion channels, transcription factors, or protein-protein interactions. Thirdly, we did not publish the in vivo efficacy of these molecules in disease models (this data is still proprietary as we found a cool indication which will be the subject of another paper). And finally, computational chemistry groups working in this area will require a broad toolkit of other deep learning systems and filtering mechanisms to go after more difficult targets with limited information available. When working backwards from a clinical trial to improve the probability of getting the molecules on the market, vast amounts of data will be required in order to train the predictive systems used for filtering compounds. It will take a few years. AI-powered drug discovery scientists will need to embrace the MMA principles; combine many strategies and styles in order to develop systems that will significantly accelerate small molecule drug discovery. There are many skeptics, who spent decades in pharma and saw hundreds of clinical failures and only few successes. Biology is very complex, chemistry is complex, and clinical trials are complex so it is very difficult to be good in all of these. But it does not mean we should not carry on and remain optimistic.
In the future the authors plan to publish a range of papers describing the molecules designed using a different form of artificial intelligence integrated into a comprehensive and fully-automated pipeline hitting very difficult targets with limited or no training sets.
Reference to the paper:
Link to the paper:
https://www.nature.com/articles/s41587-019-0224-x
The authors asked several scientists familiar with the paper to provide their comments:
“This paper is certainly a really impressive advance and likely to be applicable to many other problems in drug-design. Based on state-of-the-art reinforcement learning, I am also very impressed by the breadth of this study involving as it does molecular modeling, affinity measurements, and animal studies,” said Dr. Michael Levitt, professor of structural biology, Stanford University. Dr. Levitt received the Nobel Prize in Chemistry in 2013.
"I interacted with many AI startups in the past and Insilico was the only deep learning company with impressive, demonstrated capabilities integrating target identification and small molecule discovery. They did a lot of theoretical work in GANs from the very beginning and this experimental validation is a significant demonstration that this technology may improve and accelerate drug discovery," said Dr. John Baldoni, CTO of a stealth AI-powered drug development startup and former SVP of Platform Technology and Science at GSK.
“The generative tensorial reinforcement learning in this paper substantially advances the efficiency of biochemistry implementation in drug discovery. Yet to be further experimented at scale, this method signals a breakthrough of pharmaceutical artificial intelligence at industrial level, and may bring significant social and economic impact to our society,” said Dr. Kai-Fu Lee, founder of Sinovation Ventures, former executive of Microsoft and Google, and the original inventor of multiple AI technologies.
"I met Alex when working at OpenAI and have been excited to see him pioneer the use of GANs/RL for the pharmaceutical industry since 2016. One major criticism of GANs is that their usefulness has been limited to image editing applications, so I'm glad that Alex and his team are finding ways to use them for molecular generation," said Dr. Ian Goodfellow, the original inventor of Generative Adversarial Networks (GANs)
"This technology builds on our early work on adversarial and generative neural networks since 1990. Insilico has been working on generative models for drug discovery since 2015, and I am happy to see that their GENTRL system produced molecules that were experimentally validated in cells and in mice. AI will have a transformative effect on the pharmaceutical industry, and we need more experimental validation results to accelerate progress," said Dr. Jürgen Schmidhuber, a professor at IDSIA, co-founder of NNAISENSE, and the original inventor of many core techniques and initial concepts in the field of artificial intelligence.
“Reduction of cycle time and overall cost of goods is critical to the future success of Pharma drug discovery activities. In this paper, Insilico highlight a novel AI based technology (GAN-RL) which allowed them to identify lead molecules with efficacy in animal models in notably short timeframes. If this technology proves broadly useful it may well have transformational potential for future lead generation efforts,” said Dr. Stevan Djuric, Adjunct Professor, School of Pharmacy, High Point University and former Vice President, Discovery Chemistry and Technology, Abbvie.
“Much hyperbole exists about the promise of artificial intelligence (AI) in improving medical care and in the development of new medical tools. Here however is a paper “Deep learning enables rapid identification of potent DDR1 kinase inhibitors” recently published in Nature Biotechnology that describes an application of AI in drug discovery that is indeed important. A new drug candidate was proposed and tested preclinically in a remarkably short period of time. The results are significant for two reasons. The AI procedures replaced the role normally played by medicinal chemists, and these individuals are in limited supply. The acceleration in rate translates into longer patent coverage that improves the economics of drug development. If this approach can be generalized it could become a widely adopted method in the pharmaceutical industry,” said Dr. Charles Cantor, a professor at Boston University, co-founder of Retrotope, Inc, and former Chief Scientist of the Human Genome Project with the US Department of Energy.
Alex Zhavoronkov, PhD, is the founder and CEO of Insilico Medicine (insilico.com), a leading clinical-stage biotechnology company developing next-generation artificial intelligence and robotics platforms for drug discovery. Under his leadership Insilico raised over $400 million in multiple rounds from expert investors, opened R&D centers in six countries or regions, and partnered with multiple pharmaceutical, biotechnology, and academic institutions, nominated 8 preclinical candidates, and entered human clinical trials with AI-discovered novel target and AI-designed novel molecule.
He is also the founder and of Deep Longevity, Inc, a spin-off of Insilico Medicine developing a broad range of artificial intelligence-based biomarkers of aging and longevity servicing healthcare providers and life insurance industry. In 2020 Deep Longevity was acquired by Endurance Longevity (HK: 0575).
Since 2014 he invented critical technologies in the field of generative adversarial networks (GANs) and reinforcement learning (RL) for generation of the novel molecular structures with the desired properties and generation of synthetic biological and patient data. He also pioneered the applications of deep learning technologies for prediction of human biological age using multiple data types, transfer learning from aging into disease, target identification, and signaling pathway modeling. Prior to founding Insilico, he worked in senior roles at ATI Technologies (GPU company acquired by AMD in 2006), NeuroG Neuroinformatics, Biogerontology Research Foundation. Since 2012 he published over 160 peer-reviewed research papers, and 2 books including “The Ageless Generation: How Biomedical Advances Will Transform the Global Economy” (Macmillan, 2013). He serves on the advisory or editorial boards of Trends in Molecular Medicine, Aging Research Reviews, Aging, Frontiers in Genetics, and founded and co-chairs the Annual Aging Research, Drug Discovery and AI Forum (9th annual in 2022), the world's largest event on aging in the pharmaceutical industry. He did his two bachelor degrees at Queen’s University in Canada, masters in biotechnology at Johns Hopkins, and PhD in biophysics at MSU. He is the adjunct professor of artificial intelligence at the Buck Institute for Research on Aging.
Follow the Topic
-
Nature Biotechnology
A monthly journal covering the science and business of biotechnology, with new concepts in technology/methodology of relevance to the biological, biomedical, agricultural and environmental sciences.


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in