Predicting multiple conformations via sequence clustering and AlphaFold2
Published in Cell & Molecular Biology
Explore the Research

Predicting multiple conformations via sequence clustering and AlphaFold2 - Nature
Nature - Predicting multiple conformations via sequence clustering and AlphaFold2
The year is 2023, which means that this year we had the women’s world cup in soccer. When I’m watching a soccer game, if I were to take a picture of the field, 99% of the time that picture wouldn’t tell me anything about the outcome of the game. (Admittedly, I don't know much about soccer.) From a picture of the field, I can tell that there’s a soccer game going on, I can see that the US is playing the Netherlands, but I don’t know how many goals have been attempted or even what the score is.
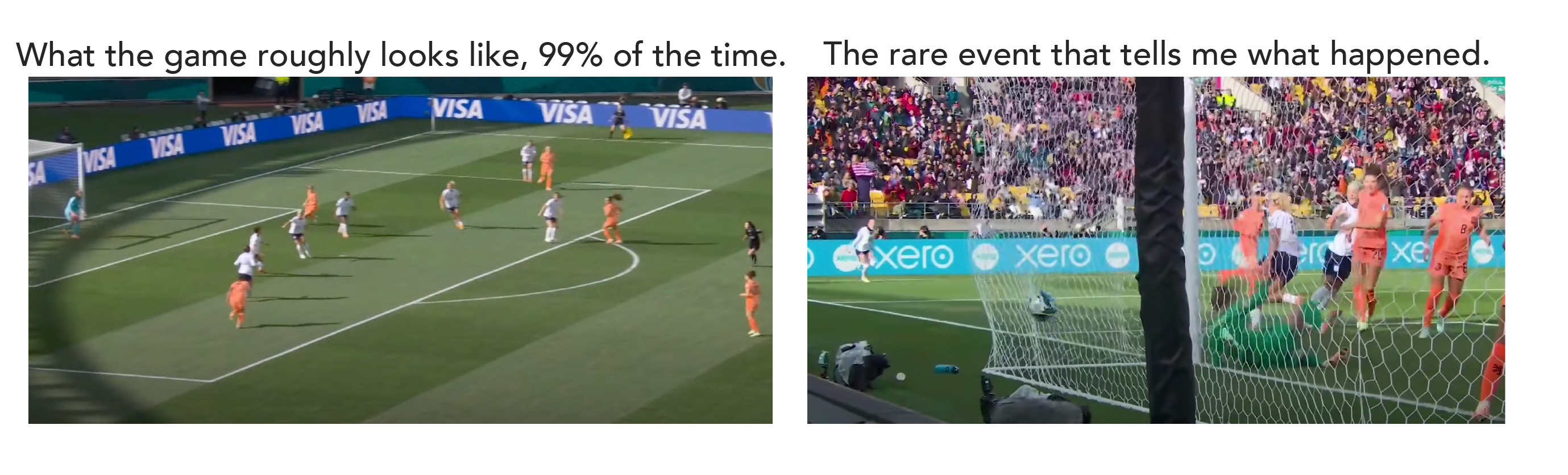
If I happened to snap a picture when US captain Lindsey Horan is going in for a header – that might tell me more about the outcome of the game. But I really need to see a picture of a ball in the Netherlands’ goal to know the US scored.
The same is true when we’re trying to understand what’s going on in biology. Single structures of macromolecules only give us snapshots of what’s going on. The Kern lab and others have repeatedly shown that not the most populated states, but rather the “rare events”, the excited states, are the crucial conformations for many biological processes, including enzyme catalysis, signaling, ligand binding, protein/protein interactions, and even drug binding.
AlphaFold2 (AF2)1 has revolutionized predicting the snapshots of the field 99% of the time – i.e., the states of proteins populated in crystal structures. AF2 built on decades of research using statistical methods to extract evolutionary couplings between amino acids from protein sequencing data, which in part reflect 3D structures. But, proteins evolve while occupying multiple conformational states! Given the essential role of these high energy conformational substates, the Kern lab immediately aimed to try to advance AF2 to predict those other states.
We needed an idea of how do it, and a system to test the idea. The idea was that the conformational substates are under evolutionary selection due to their essential roles in biological function, therefore signals for these other conformations should be in the evolutionary conservation within the sequences. In fact, 10 years ago, researchers pointed out that they could detect evolutionary couplings for multiple conformations of ion channels in evolutionary data.2
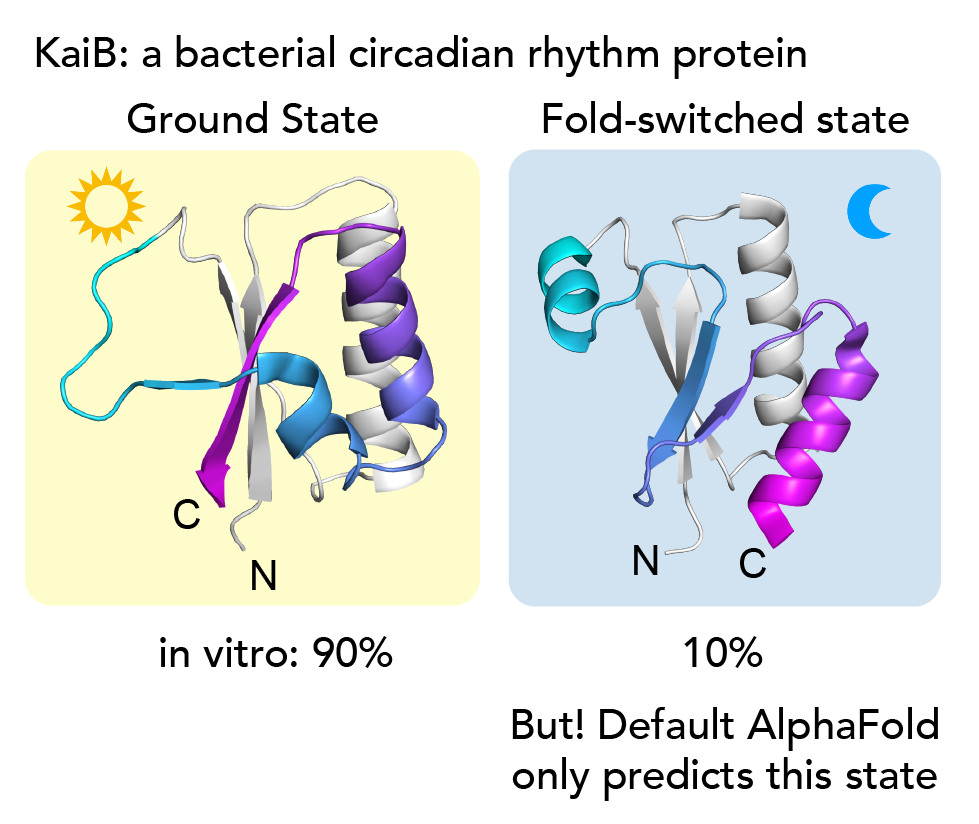
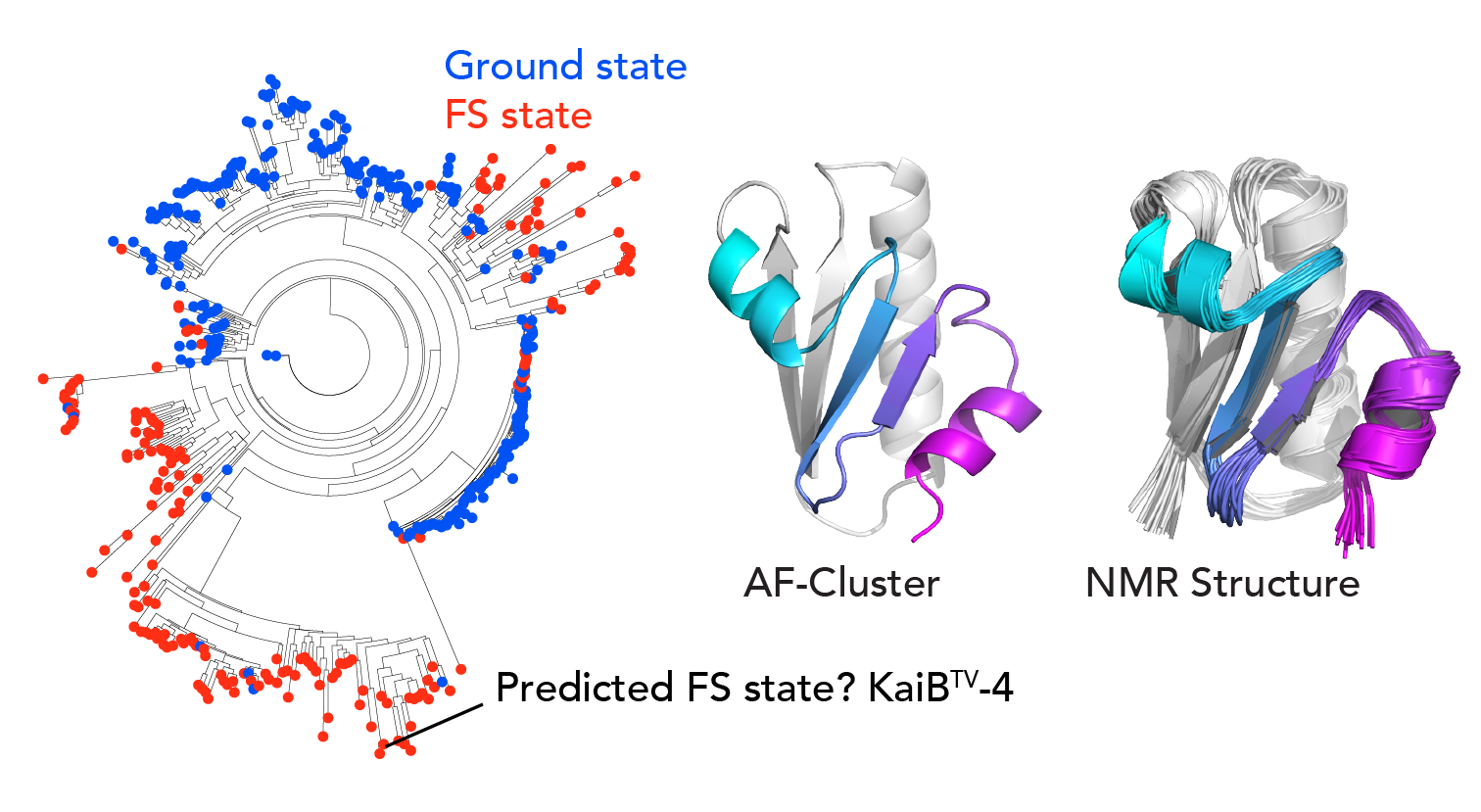
As a system to investigate our idea, we chose the little protein KaiB - 90 amino acids long- that we study in the lab. It undergoes a very dramatic conformational change, but none of the existing AI methods could predict both states. KaiB conformational switching is involved in regulating the circadian rhythms of cyanobacteria3,4. With NMR spectroscopy, we observe that our KaiB variant occupies a “Ground state” and a minor “Fold-switched” (FS) state in equilibrium, with the minor state being occupied to about 10%. The C-terminal half of the protein undergoes a dramatic conformational change where beta strands convert into helices and helices into strands (see above). Only the FS state can bind KaiB's partner, KaiC. Intriguingly, no matter what lab members tried in current variations of AF2, they only ever got predictions for the FS state. While experimenting with AF2 one day, I realized that if I used only the closest 50 sequences to KaiB by phylogenetic distance, that returned the ground state. But, if I took the closest 100 sequences, the prediction flipped back to the FS state!
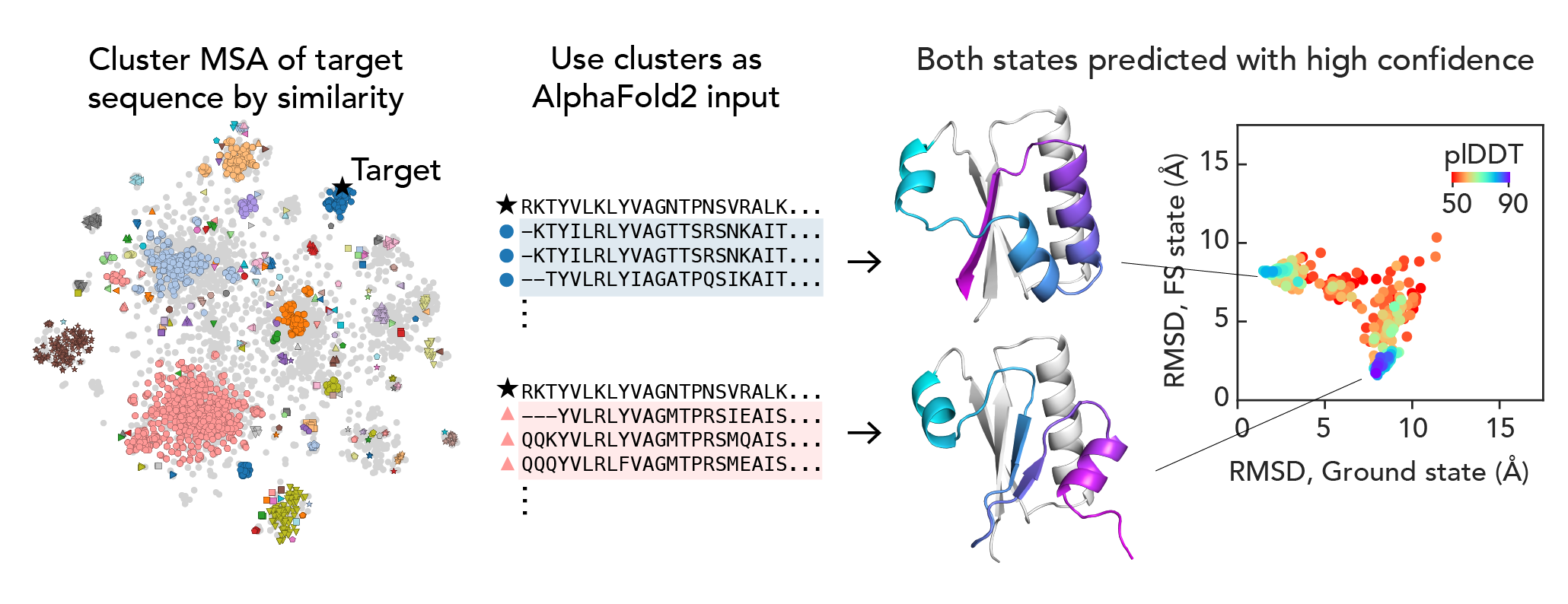
We wondered if there were pockets of local evolutionary signal for one or the other state across the KaiB family. We found that if we clustered the input MSA by sequence, a simple way of getting at evolutionary similarity, and used those clusters as input MSAs for AF2, we got a distribution of structures, and the highest-scored structures were exactly the two states that we know to be true!
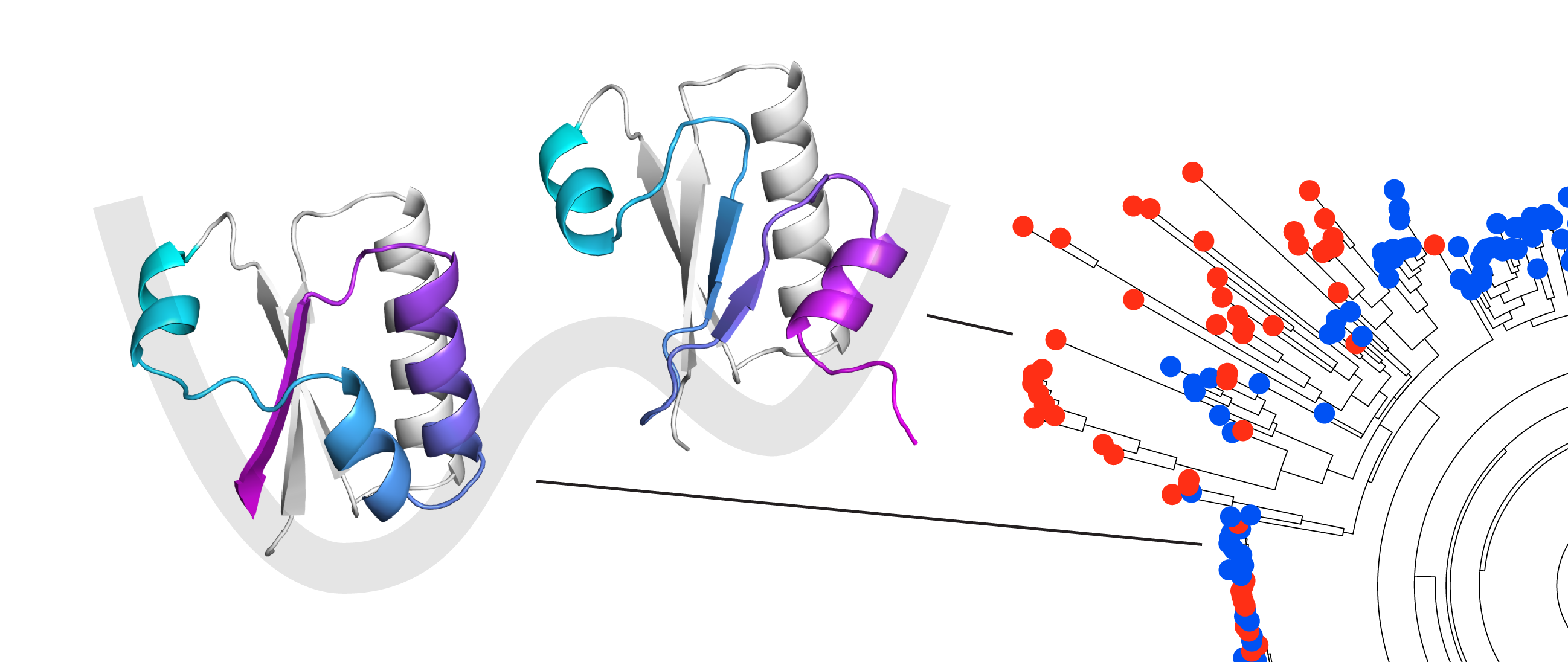
Our next question was, where was this signal at the sequence level for the excited state coming from? We created a phylogenetic tree for KaiB and made predictions with “shallow MSAs” across it. We saw that there were pockets of predictions for one state or the other state. We noticed that for some variants that were evolutionarily close to the well-studied KaiB variant in cyanobacteria, AF-Cluster predicted was stabilized for the other state. We felt the strong need to experimentally test our AF-cluster method and characterized one of these with NMR, and sure enough, the prediction was correct – it was stabilized for the other state.
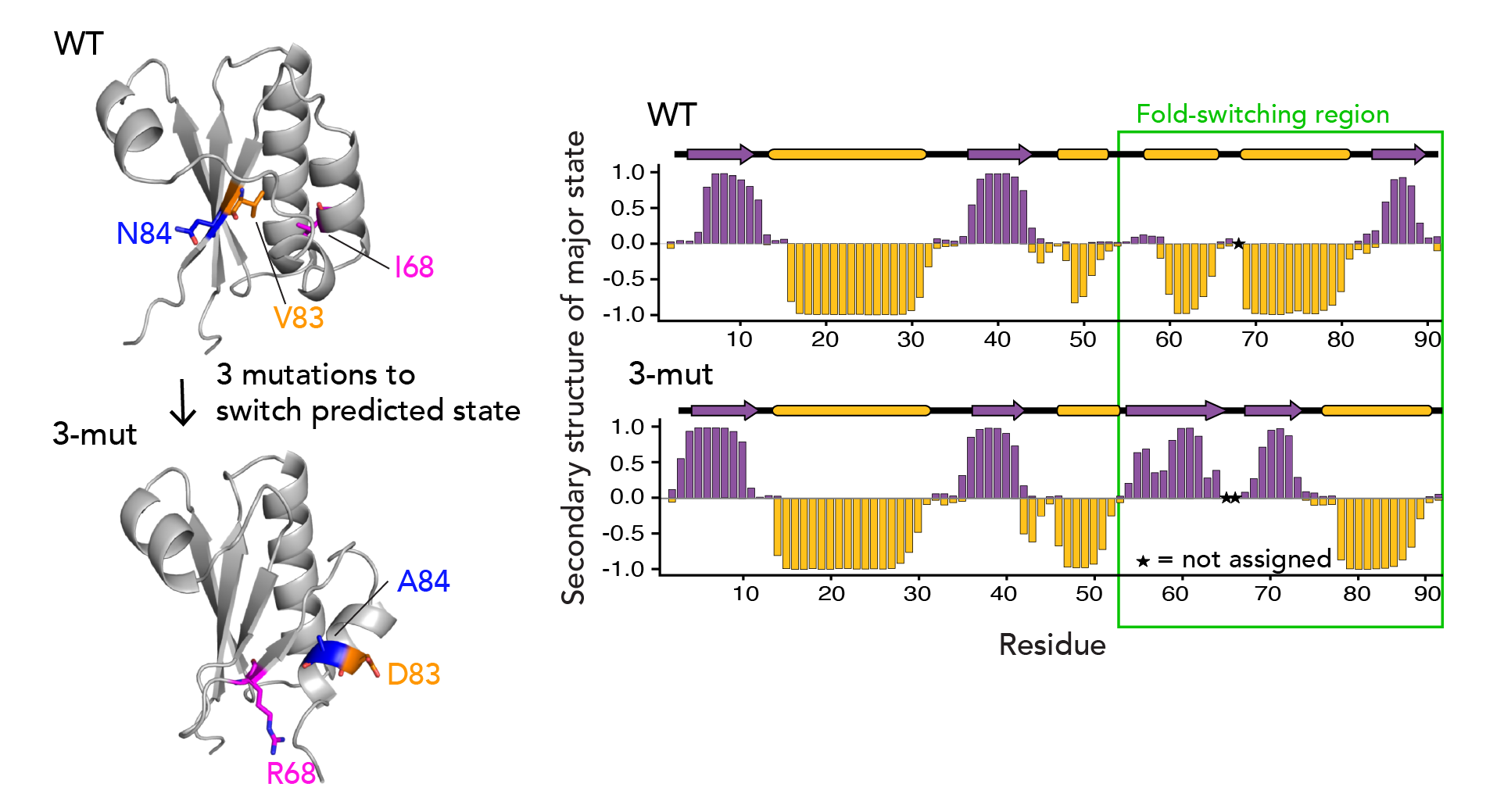
There’s been immense interest if AF2 can predict the effects of point mutations, such as cancer-causing mutations. Accordingly, we were curious what the sensitivity of AF-Cluster was to changes in point mutations in our system. We found that we needed just 3 point mutations to change AF-clusters’s prediction of the ground state for KaiB to the FS state with high confidence. Strikingly, when we characterized this triple mutant with NMR, we found that it indeed had flipped its equilibrium to favor the FS state (see below).
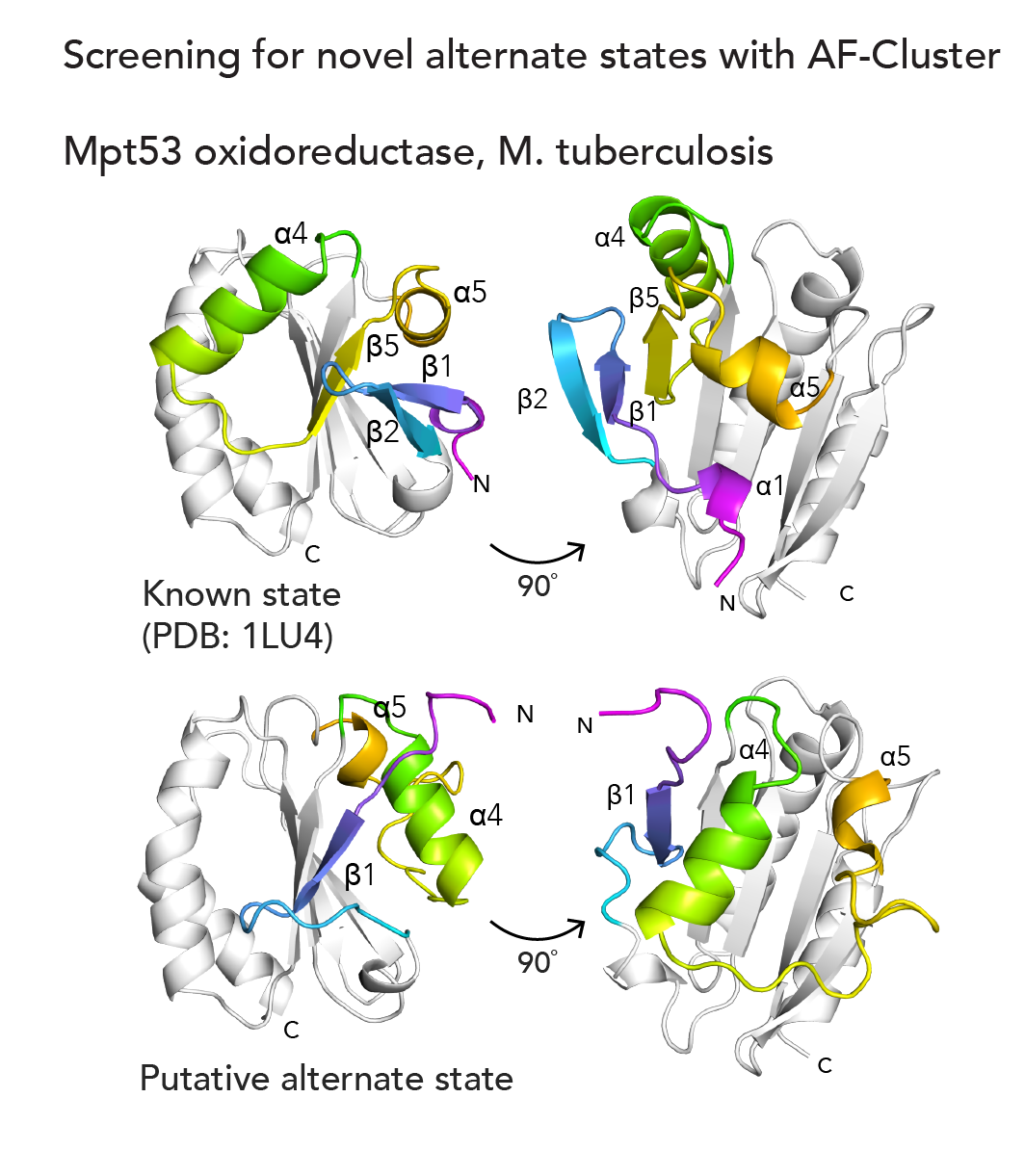
The next question we tackled was, could we use AF-Cluster to computationally screen for previously undiscovered alternate states in other protein families? Indeed, in a small screen of ~600 protein families from an existing database, we identified an alternate novel state for Mpt53, a secreted oxidoreductase in M. tuberculosis5 (below). We’re really excited about the potential AF-Cluster and related methods hold for the ability to discover new biology.
AF-Cluster still leaves much room for improvement for modeling conformational landscapes. For one, the relative free energies of states cannot be determined reliably, nor the rates and mechanisms for transitioning between them.
Going back to soccer, the fastest way to know the outcome of a game is to watch a highlight reel – video sampled to show the 1% of the time when there were shots on goal, i.e. the excited states. How far away are biology highlight reels? At least hopefully now we’re a little closer.
References
1 Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583-589 (2021).
2 Hopf, T. A. et al. Three-dimensional structures of membrane proteins from genomic sequencing. Cell 149, 1607-1621 (2012).
3 Chang, Y. G. et al. Circadian rhythms. A protein fold switch joins the circadian oscillator to clock output in cyanobacteria. Science 349, 324-328 (2015).
4 Pitsawong, W. et al. From primordial clocks to circadian oscillators. Nature (2023).
5 Wang, L. et al. Oxidization of TGFbeta-activated kinase by MPT53 is required for immunity to Mycobacterium tuberculosis. Nat Microbiol 4, 1378-1388 (2019).
Follow the Topic
-
Nature
A weekly international journal publishing the finest peer-reviewed research in all fields of science and technology on the basis of its originality, importance, interdisciplinary interest, timeliness, accessibility, elegance and surprising conclusions.

Related Collections
With Collections, you can get published faster and increase your visibility.
Carbon Dioxide Removal
Publishing Model: Hybrid
Deadline: Jan 16, 2027

Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in