Re-sensitizing CML to Imatinib: Dual Targeting of HDACs and BCR-ABL with Martinostat
Published in Cancer, General & Internal Medicine, and Pharmacy & Pharmacology
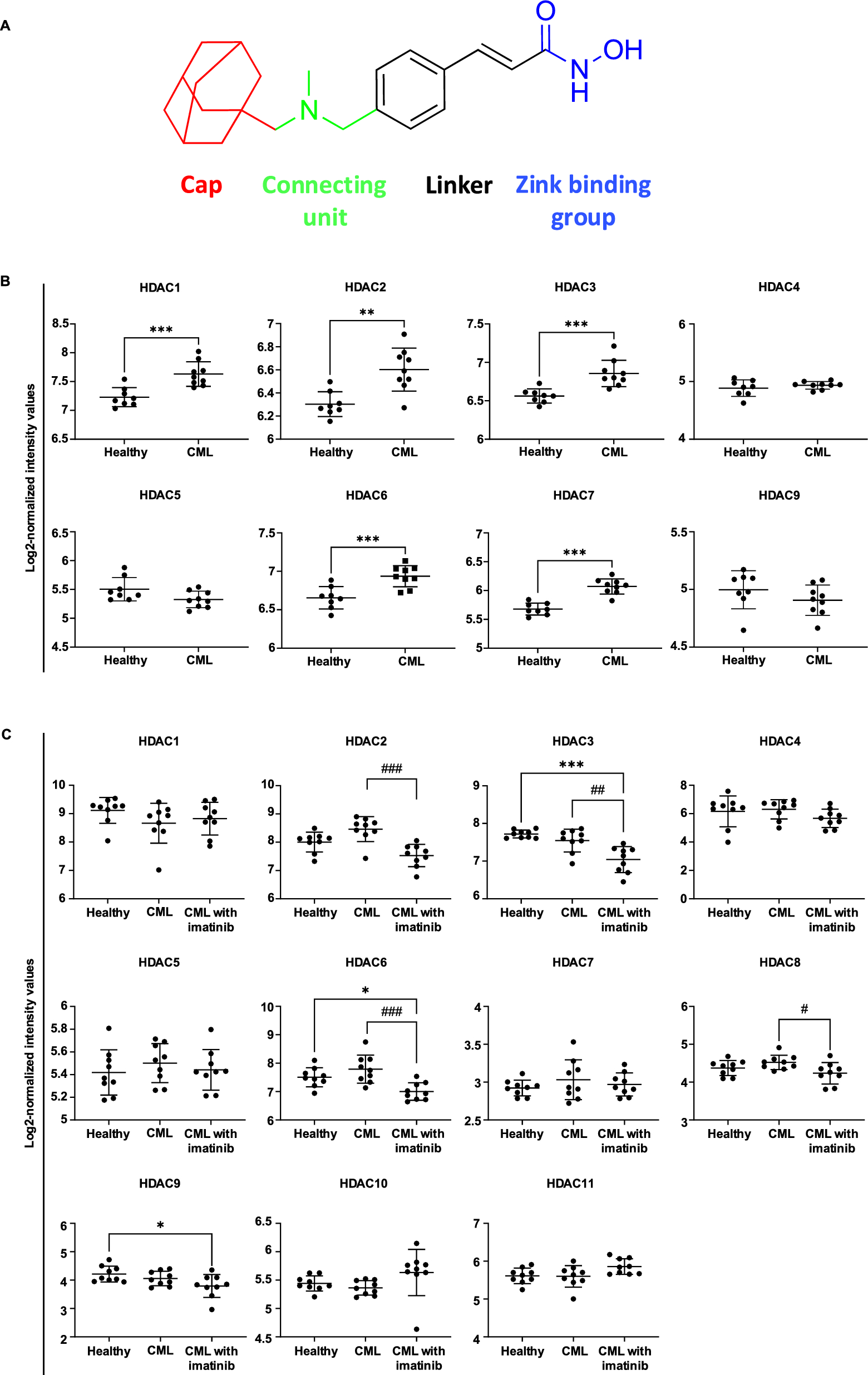

Chronic Myeloid Leukemia (CML) is a hematological malignancy that accounts for 15% of adult leukemia cases, driven primarily by the BCR-ABL oncoprotein. Although tyrosine kinase inhibitors (TKIs) like imatinib have revolutionized CML treatment, resistance to TKIs has become a therapeutic challenge. Histone deacetylases (HDACs) play a crucial role in epigenetic modulation and are frequently upregulated in cancers, including leukemia [1-4]. In this study, we present the first demonstration of the potent anti-leukemic activity of the HDAC inhibitor martinostat in both TKI-sensitive and -resistant CML [5].
In previous studies, martinostat was identified as a specific inhibitor of HDAC class I enzymes by targeting overexpressed HDAC class I in neurological disorders [6] and prostate cancer [7]. However, we first identified martinostat as a pan-HDAC inhibitor that significantly targets HDAC classes I, II, and IV compared to the clinically approved HDAC inhibitor SAHA. Martinostat exhibited favorable drug-like properties aligned with Lipinski’s rule of five, supporting its suitability as a therapeutic candidate. Additionally, we investigated the anti-leukemic potential of martinostat in CML cell models. Martinostat selectively reduced the proliferation and viability of CML cells and induced apoptosis across multiple CML models, including imatinib-resistant cell lines and patient-derived blasts, while showing minimal cytotoxicity toward healthy cells and low developmental toxicity in zebrafish embryos.
Martinostat induced apoptosis by activating caspase pathways and promoting immunogenic cell death through HMGB1 release. Transmission electron microscopy (TEM) observations indicated morphological changes and increased autophagic activity following martinostat treatment. In mRNA-seq analysis, martinostat treatment significantly impacted key gene pathways related to leukemia cell survival and proliferation. These findings collectively support martinostat’s potential as a therapeutic agent for CML.
Regarding the HDAC inhibitory effect, martinostat induced histone acetylation at significantly lower concentrations than SAHA, indicating enhanced potency in epigenetic modulation. Compared to SAHA, martinostat more efficiently suppressed leukemic colony formation, elicited morphological alterations, and modified cell distribution and proliferative capacity. Computational docking studies revealed that martinostat interacts with the binding pocket of HDAC isoenzymes in a similar manner to SAHA; however, in detailed docking simulations, martinostat exhibited notably stronger binding affinity for selected HDAC isoenzymes than SAHA, reinforcing its potential as a more effective HDAC inhibitor.
In combination with TKI imatinib, martinostat exhibited a synergistic effect in both imatinib-sensitive and resistant CML cells. Against the BCR-ABL molecular pathway, the primary cause of CML, the combination of martinostat and imatinib abolished the activation of the BCR-ABL and STAT5 pathways by overcoming TKI resistance and inducing cell death compared to treatment with either martinostat or imatinib alone. This synergy was validated in vivo using a mouse xenograft model, as evidenced by significantly inhibited tumor growth and reduced tumor volume while causing no organ damage in the mice.
Collectively, these findings underscore martinostat's potential as a selective, low-toxicity HDACi with notable anti-cancer effects in CML. To address TKI resistance, martinostat emerges as a promising therapeutic candidate in combination with imatinib, potentially offering an effective strategy for enhancing CML treatment outcomes.
.png)
A mechanistic overview describing the effect of Martinostat.
Created in BioRender. Yang, H and Diederich, M. (2025)
References:
1. Losson H, Schnekenburger M, Dicato M, Diederich M: HDAC6-an Emerging Target Against Chronic Myeloid Leukemia? Cancers (Basel) 2020, 12(2).
2. Losson H, Gajulapalli SR, Lernoux M, Lee JY, Mazumder A, Gerard D, Seidel C, Hahn H, Christov C, Dicato M et al: The HDAC6 inhibitor 7b induces BCR-ABL ubiquitination and downregulation and synergizes with imatinib to trigger apoptosis in chronic myeloid leukemia. Pharmacol Res 2020, 160:105058.
3. Lernoux M, Schnekenburger M, Losson H, Vermeulen K, Hahn H, Gerard D, Lee JY, Mazumder A, Ahamed M, Christov C et al: Novel HDAC inhibitor MAKV-8 and imatinib synergistically kill chronic myeloid leukemia cells via inhibition of BCR-ABL/MYC-signaling: effect on imatinib resistance and stem cells. Clin Epigenetics 2020, 12(1):69.
4. Lernoux M, Schnekenburger M, Dicato M, Diederich M: Epigenetic mechanisms underlying the therapeutic effects of HDAC inhibitors in chronic myeloid leukemia. Biochem Pharmacol 2020, 173:113698.
5. Yang H, Li V, Park SJ, Cheon SW, Lorant A, Mazumder A, Lee JY, Orlikova-Boyer B, Cerella C, Christov C et al: Martinostat as a novel HDAC inhibitor to overcome tyrosine kinase inhibitor resistance in chronic myeloid leukemia. Clinical Epigenetics 2025, 17(1):125.
6. Pascoal TA, Chamoun M, Lax E, Wey HY, Shin M, Ng KP, Kang MS, Mathotaarachchi S, Benedet AL, Therriault J et al: [(11)C]Martinostat PET analysis reveals reduced HDAC I availability in Alzheimer's disease. Nat Commun 2022, 13(1):4171.
7. Chen Z, Wang X, Yang X, Xu Y, Yang Y, Wang H, Li T, Bai P, Yuan G, Chen H et al: Imaging assisted evaluation of antitumor efficacy of a new histone deacetylase inhibitor in the castration-resistant prostate cancer. European Journal of Nuclear Medicine and Molecular Imaging 2021, 48(1):53-66.
Follow the Topic
-
Clinical Epigenetics
Encompassing the broad spectrum of epigenetics research from basic research to innovations in therapeutic treatments, this is a top tier, open access journal devoted to the study of epigenetic principles and mechanisms as applied to human development, disease, diagnosis and treatment.

Related Collections
With Collections, you can get published faster and increase your visibility.
Artificial Intelligence and Advanced Technologies in Clinical Epigenetics
Clinical epigenetics investigates how epigenetic modifications, such as DNA methylation, histone modifications, and chromatin remodeling, shape disease mechanisms, influence patient outcomes, and guide therapeutic strategies. These changes often reflect environmental exposures and pathological states, making them critical biomarkers and promising targets for precision medicine.
Recent advances in high-resolution “omics” technologies enable detailed mapping of epigenetic landscapes in clinical samples. Integrative network-based approaches provide systems-level insights into disrupted signaling pathways, while artificial intelligence (AI) and machine learning (ML) approaches are emerging as transformative tools for analyzing complex epigenetic datasets, predicting disease risk, and informing personalized interventions.
This Collection seeks to highlight cutting-edge research at the intersection of AI, ML, systems biology, and computational modeling with clinical applications in epigenetics. We welcome studies that leverage innovative technologies, AI-driven discovery, and translational strategies to improve diagnosis, prognosis, and treatment. By emphasizing interdisciplinary advances, this Collection aims to accelerate the integration of epigenetic insights into clinical practice.
Topics of interest (but not limited to):
• AI-driven epigenetic biomarker discovery for disease diagnosis and prognosis
• Integration of multi-omics data using ML and/or network-based approaches in clinical settings
• Predictive modeling of treatment response through epigenetic signatures and computational frameworks
• Development of clinical decision-support tools leveraging epigenetic data and artificial intelligence
• Translational applications of epigenetic technologies for personalized medicine and therapeutic targeting
All submissions in this collection undergo the journal’s standard peer review process. Similarly, all manuscripts authored by a Guest Editor(s) will be handled by the Editor-in-Chief. As an open access publication, this journal levies an article processing fee (details here). We recognize that many key stakeholders may not have access to such resources and are committed to supporting participation in this issue wherever resources are a barrier. For more information about what support may be available, please visit OA funding and support, or email OAfundingpolicy@springernature.com or the Editor-in-Chief.
Publishing Model: Open Access
Deadline: Oct 12, 2026
Pollution and Epigenetics
From diesel particulates to endocrine disruptors, asbestos, heavy metals to molecules like bisphenol A (BPA), it is becoming increasingly clear that man’s propensity to pollute has significant consequences on human health. Moreover, strong evidence now links such pollution to changes within our epigenomes. In this new thematic series in Clinical Epigenetics, we explore the causes and consequences of pollution on the epigenome, how this may have effects not only on the epigenetics of the individual exposed to such pollution, but also review how this may be further exacerbated by downstream or “transgenerational” inheritance of these epigenetic changes.
Publishing Model: Open Access
Deadline: Ongoing


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in