Settling the matter of the role of vibrations in the stability of high-entropy carbides
Published in Electrical & Electronic Engineering
High-entropy ceramics have received considerable attention due to their exceptional chemical stability and physical properties. They typically consist of a chemically disordered cation site with four or more species and a chemically ordered anion site. Computational assessment of the stability of these materials typically relies on using enthalpies from density functional theory (DFT) along with the ideal configurational entropy, simplified descriptors, or machine learning. The extent of vibrational contributions (phonons) to the stability of these materials, however, remains elusive. Vibrations have so far only been investigated in high-entropy alloys, which lack ordered sites and thus have different atomic environments. Research in these alloys have produced controversial results about the extent of vibrational contributions. In ceramics, phonon calculations are often avoided due to high computational costs and complexity. We wanted to shed light on this irresolution and determine the role of vibrations for the stability of high-entropy ceramics.
The biggest challenge in modeling disordered materials using first-principles methods is the appropriate description of the structure as standard DFT requires ordered representations of the disordered materials. Research in high-entropy alloys has used special quasi-random structures that use large supercells with the “most random” atom configuration. This corresponds to an infinite-temperature solution, which may not be appropriate for finite temperatures. At AFLOW, we had developed the Partical OCCupation (POCC) module to mitigate this issue. It describes disordered materials using an ensemble of smaller, ordered representatives (Figure 1a) that are weighted according to Boltzmann statistics. Our strategy was to combine the Automatic Phonon Library (APL), also developed by us at AFLOW, with POCC to determine the vibrational properties of select high-entropy carbides. We used the high-entropy carbides (HfNbTaTiR)C5 (R = V, W, Zr, HEC-R for short) as a testbed. These materials are known to form single-phase rock-salt materials.
Since an accurate description of the structure is fundamental to calculating phonon properties, we were first interested to see if AFLOW-POCC provides a reasonable representation of the disordered material. The average lattice parameters of the ordered representatives were in excellent agreement with experiments. We also looked at the interatomic distances and found that the carbon atoms were displaced from their ideal positions whereas the metals were more evenly spaced. The origin of these displacements lies in the competition between enthalpy and entropy. To maximize configurational entropy, we would expect to find identical metal-carbon distance throughout the crystal and thus the most uniform atomic environments. However, this would incur enthalpic penalties since every metal has a different ideal metal-carbon distance. The result is that the carbon atoms get displaced to provide nearest-neighbor environments that are as uniform as possible without straying too far from the equilibrium metal-carbon distances. AFLOW-POCC thus provides structures that are consistent with experiments and physical and chemical principles, which encouraged us to integrate APL into POCC.
We then performed phonon calculations to calculate the phonon density of states (pDOS) for each ordered representative and thermally weighted each structure for the ensemble-averaged pDOS (Figure 1b). Some derivative structures were dynamically unstable. Treating those would require prohibitively expensive methods, so we decided to exclude these structures from the ensemble, believing the overall conclusions not to be substantially affected. The local distortions in the atomic environments lead to force-constant disorder, which causes the pDOS for the metal and the carbon sites to broaden. For the metals, additional broadening can be observed due to mass disorder. Interestingly, only the broadening on the metal sites affects the vibrational free energy: mass disorder destabilizes the high-entropy material whereas force-constant disorder has the opposite effect. The force-constant disorder on the carbide sites has no effect on the energies.
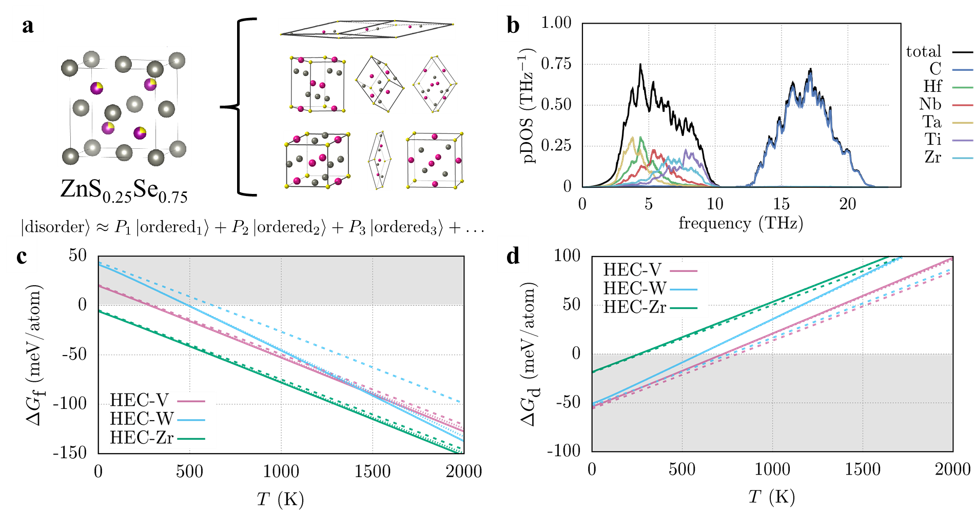
Figure 1 a Schematic representation of the Partial OCCupation (POCC) method using ZnS0.25Se0.75 as an example. The disordered material (left) is described using an ensemble of ordered representatives (right) using thermodynamic weights P. b Phonon density of states (pDOS) of (HfNbTaTiZr)C5 (HEC-Zr). c and d Gibbs free energy for the formation (DGf) and decomposition reactions (DGd) of the high-entropy carbides with (solid) and without (dashed) vibrational contributions.
At last, we were interested in how vibrations influence the stability of these high-entropy carbides. They are synthesized from their binary precursors:
HfC + NbC + TaC + TiC + RC → (HfNbTaTiR)C5,
and are predicted to decompose according to the following reactions:
(HfNbTaTiV)C5 → HfNbC2 + TaTiC2 + ⅚ V6C5 + ⅙ C,
(HfNbTaTiW)C5 → HfNbC2 + TaTiC2 +WC,
(HfNbTaTiZr)C5 → HfNbC2 + TaTiC2 + ZrC.
We calculated the Gibbs free energy for each of these reactions with and without vibrational contributions. For the formation of HEC-V and HEC-Zr (Figure 1c), vibrations contribute only little. For HEC-W, however, the difference in transition temperature is measurable (150 K). Looking at the individual vibrational free energies, we found that the reason is not an exceptionally low value for HEC-W but a higher value for WC. In contrast to the other rock-salt binary carbides, WC crystallizes in a hexagonal structure with lower symmetry. In general, low symmetry results in higher phonon frequencies and thus less negative vibrational free energies. The decomposition reaction (Figure 1d) shows similar trends: the difference in the Gibbs free energy is imperceptible for HEC-Zr and substantial for HEC-W, even though they are predicted to decompose into ternary carbides. HEC-V is in-between these compounds. A closer look at the structures of HfNbC2 and TaTiC2 revealed that nearest-neighbor atomic environments are similar to rock salt. V6C5 can also be understood as a rock-salt structure with a large vacancy concentration. We thus concluded that the nearest-neighbor environments in reactants and products are the primary factor that determines when vibrations are significant.
A difference of 150 K may not strongly affect synthesizability, but since we used the ideal configurational entropy, the changes in transition temperatures due to vibrations are likely a lower limit. Even a difference 150 K can have significant effects on phase transition behavior though. Time-temperature-transition curves of alloys and ceramics show that transition times can vary by orders of magnitude within a 150 K window. Additionally, synthesis conditions can strongly influence physical properties: sintering at high temperatures can degrade mechanical properties of high-entropy carbides. Since a material is only as good as its performance in applications, it is as critical to determine under which conditions these materials can be synthesized. Our research shows that for these purposes, vibrational contributions cannot be ignored a priori.
Still, some questions still remain. The extent of anharmonic contributions (phonon-phonon interactions) has yet to be determined and is a formidable computational challenge to overcome. If simple solid solutions can be a guide, however, anharmonicity would further stabilize the high-entropy material. High-entropy carbides also have a high tolerance for defects, which can influence the vibrational properties of these materials. Both are exciting avenues to explore in future research.
Acknowledgments
The author thanks Xiomara Compilongo for fruitful discussions. This work was supported by the United States Department of Defense Office of Naval Research (N00014-15-1-2863, N00014-21-1-2132, N00014-20-1-2525, N00014-20-1-2299), and the National Science Foundation under DMREF Grant No. DMR-1921909.
Follow the Topic
-
Nature Communications
An open access, multidisciplinary journal dedicated to publishing high-quality research in all areas of the biological, health, physical, chemical and Earth sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Women's Health
Publishing Model: Hybrid
Deadline: Ongoing
Tumor Microenvironment Crosstalk and Therapeutic Implications
Publishing Model: Hybrid
Deadline: Nov 02, 2026


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in