Systematic decomposition of sequence determinants governing CRISPR/Cas9 specificity
Published in Bioengineering & Biotechnology
The story behind this paper started in the fall of 2018 when I joined the Xu Lab at The University of Texas MD Anderson Cancer Center. At that time, Dr. Han Xu had uncovered the sequence determinants governing the efficiency of guide RNA [1] and had extensive experience in guide RNA design. Before joining his lab, I was very interested in CRISPR technology, but was not familiar with this area. He guided me into the CRISPR field and worked alongside me and my collaborator Wei He to complete our study.
CRISPR/Cas9 has been widely used for genome editing, but before the start of my work in 2018, the outcomes of Cas9-mediated double-strand break repair were not fully understood. Doench et al. [2] developed the Cutting Frequency Determination (CFD) score to predict the off-target potential of gRNAs using a library that targets the coding sequence of human CD33 with all possible sgRNAs; however, this prediction is derived from a limited number of gRNAs and is an indirect measurement of editing by Cas9. Subsequently, during the stage of research planning, we proposed two projects: (1) systematic evaluation and prediction of the mutations generated by repair of Cas9-induced double-strand breaks; (2) systematic decomposition of sequence determinants governing CRISPR/Cas9 specificity. While we were still considering leveraging a synthetic gRNA-target system to fulfill these two projects, the FORECast tool [3] to predict the outcome of an edit mediated by Cas9 using the gRNA-target system was published on bioRxiv. Afterward, the predictable and precise template-free edits by Cas9 were systematically revealed by InDelphi [4] and Lindel [5]. Thus, we reshaped our research aim and focused on the systematic decomposition of sequence-dependent rules underlying Cas9.
During lentiviral production [6] and PCR [7] in this type of multiplexed gRNA-target system, swapping is an issue that can confound data interpretation and lead to a waste of sequencing reads. This could be up to 30-50%, as seen in the above-mentioned studies predicting the mutations on on-target sequences. To obtain some sense of Cas9 off-targeting from pure datasets, we first designed 6 libraries consisting of a single gRNA paired with all its possible 1-mismatch targets, all its possible 2-mismatch targets and some random 3- to 6-mismatch targets. Among these 6 gRNAs, we found 4 gRNAs were associated with high off-target potential but the other 2 displayed mismatch intolerance. This guide-intrinsic effect was also present regardless of mismatch contexts. Moreover, we found an “epistasis-like” combinatorial effect with 2-mismatch targets, which is inconsistent with the previous theoretical model assuming the combinatorial effect of two or more mismatches to be “marginally independent” [2]. Motivated by the great curiosity of mechanisms underlying these two observations, we then went forward with newly designed libraries with multiplexed gRNAs to uncover the sequence determinants underlying Cas9 off-target effects.
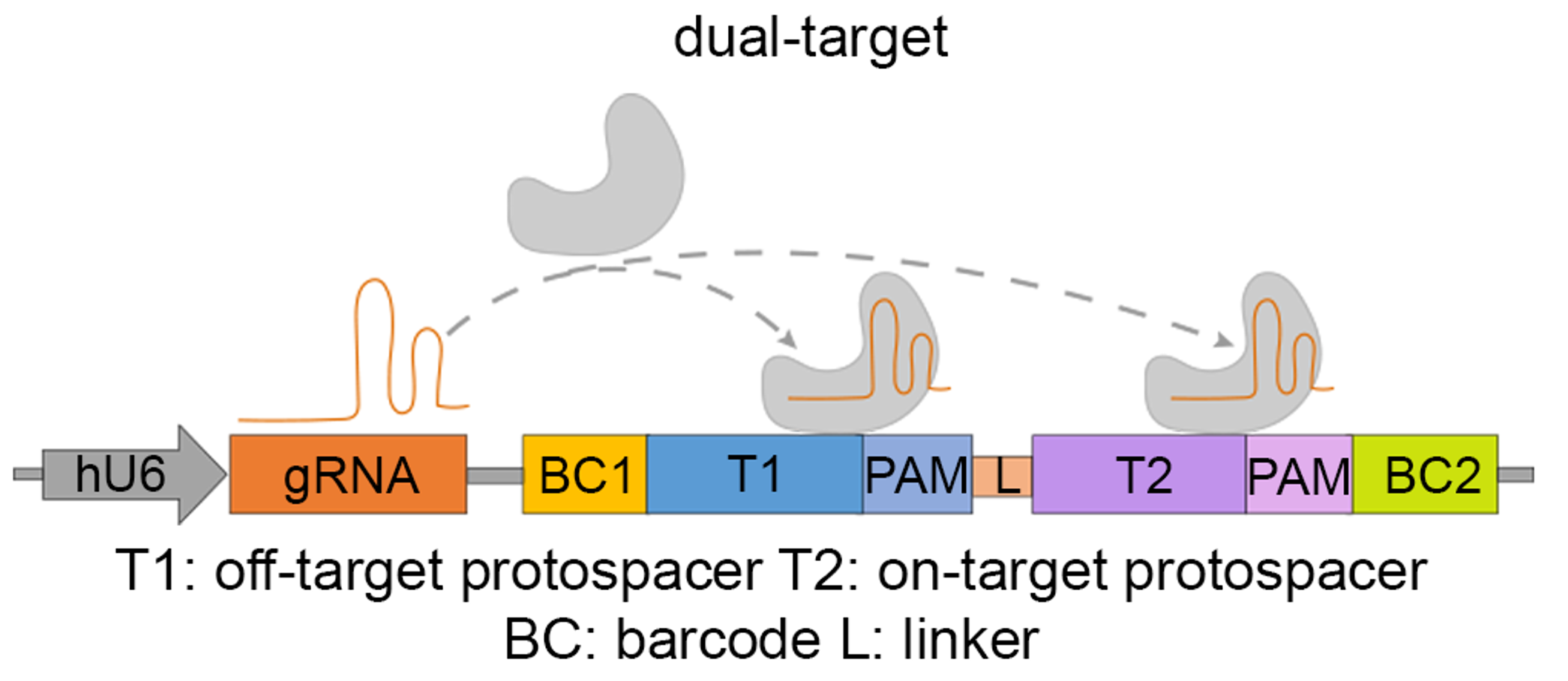
To avoid the confounding factors such as swapping and PCR biases in the multiplexed gRNA-target system, we modified the paired gRNA-target system by introducing a dual-target sequence that contains two 23-bp PAM-endowed target sequences arranged in tandem, corresponding to an off-target (left) and an on-target (right) (Figure 1). Since the off- and on-targets are integrated to the same genomic locus and are PCR-amplified together, the on-target cleavage rate acts as an internal control for the normalization against confounding factors in the experiment.
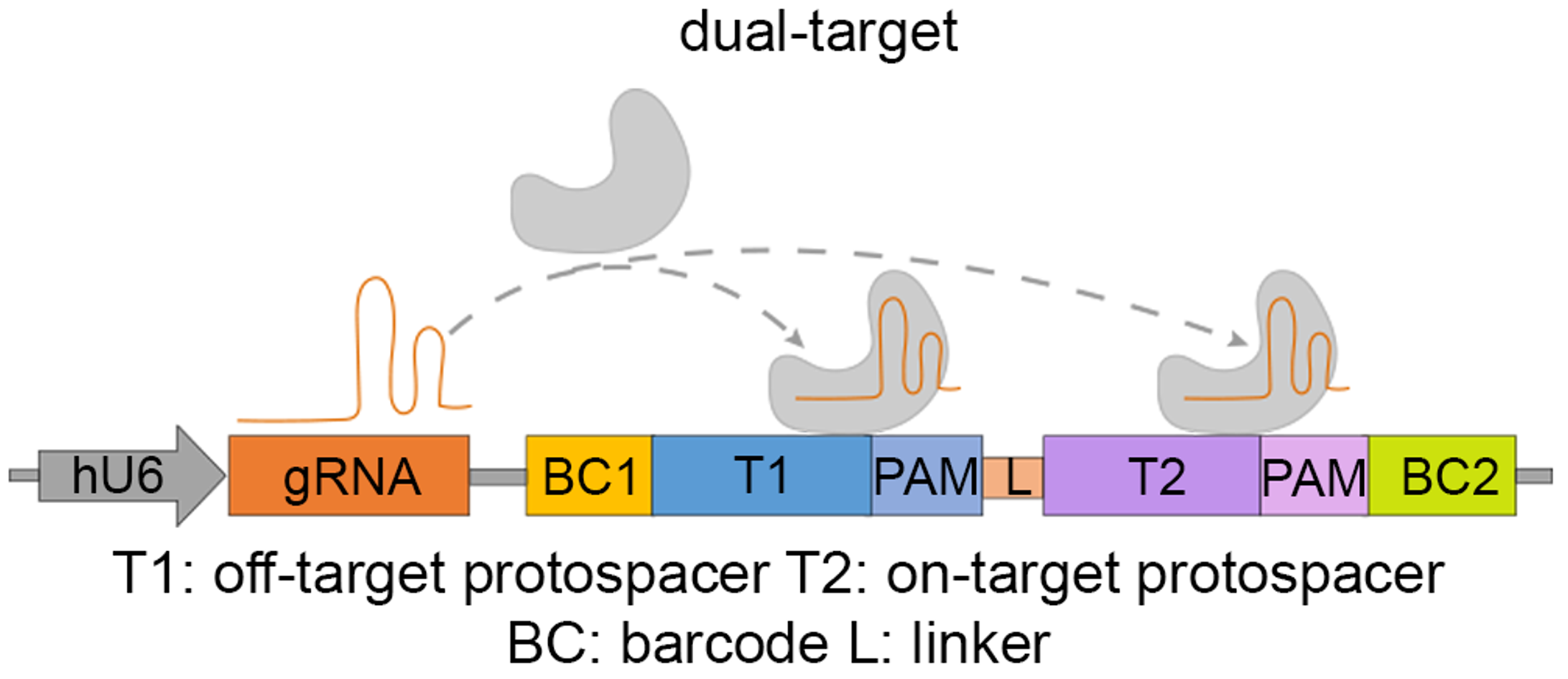
Figure 1. The design of dual-target system
We first tested the dual-target system on two gRNAs associated with distinct repair mechanisms upon double-strand breaks. To explore the editing outcomes mediated by different cleavage events at almost identical tandem targets, we designed 4 types of dual-target sequences to represent 4 combinations of cleavage events (no cleavage, left, right, and both), where the cleavage can be turned off at a specific target by the replacement of the “NGG” PAM sequences with “NTT” (Figure 2).
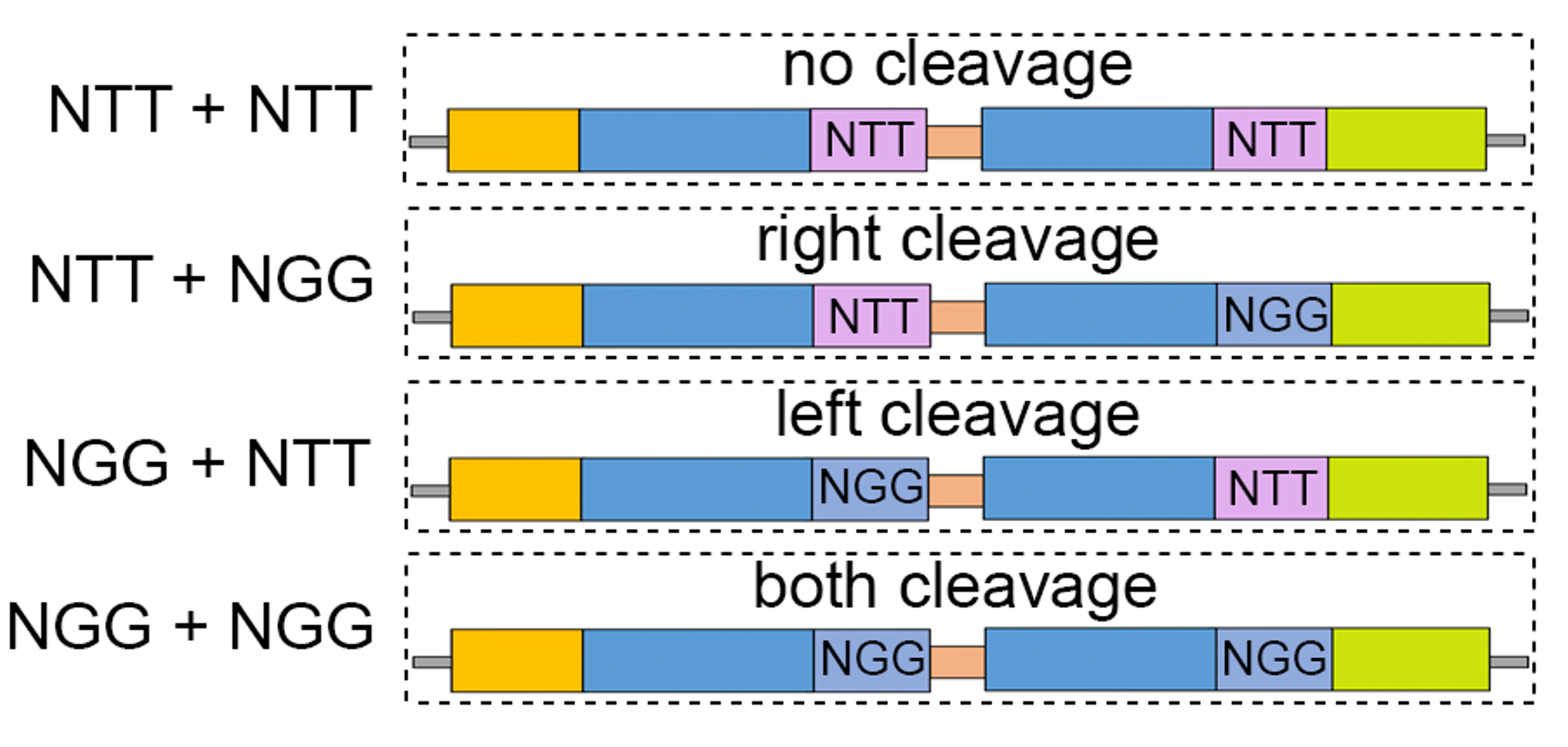
Figure 2. The design of 4 dual-target sequences corresponding to 4 cleavage types
Based on the editing outcomes of the two gRNAs, we found that, in addition to anticipated small indels, large deletions (>30-nt) were enriched when cleavage occurred at both targets (NGG + NGG) or the left target alone (NGG+NTT). The latter is likely due to the similarity of the two target sequences that induces long-range resection via microhomology-mediated end joining (MMEJ). These observations were consistent between the two gRNAs, suggesting a general cleavage-editing model as demonstrated in Figure 3.
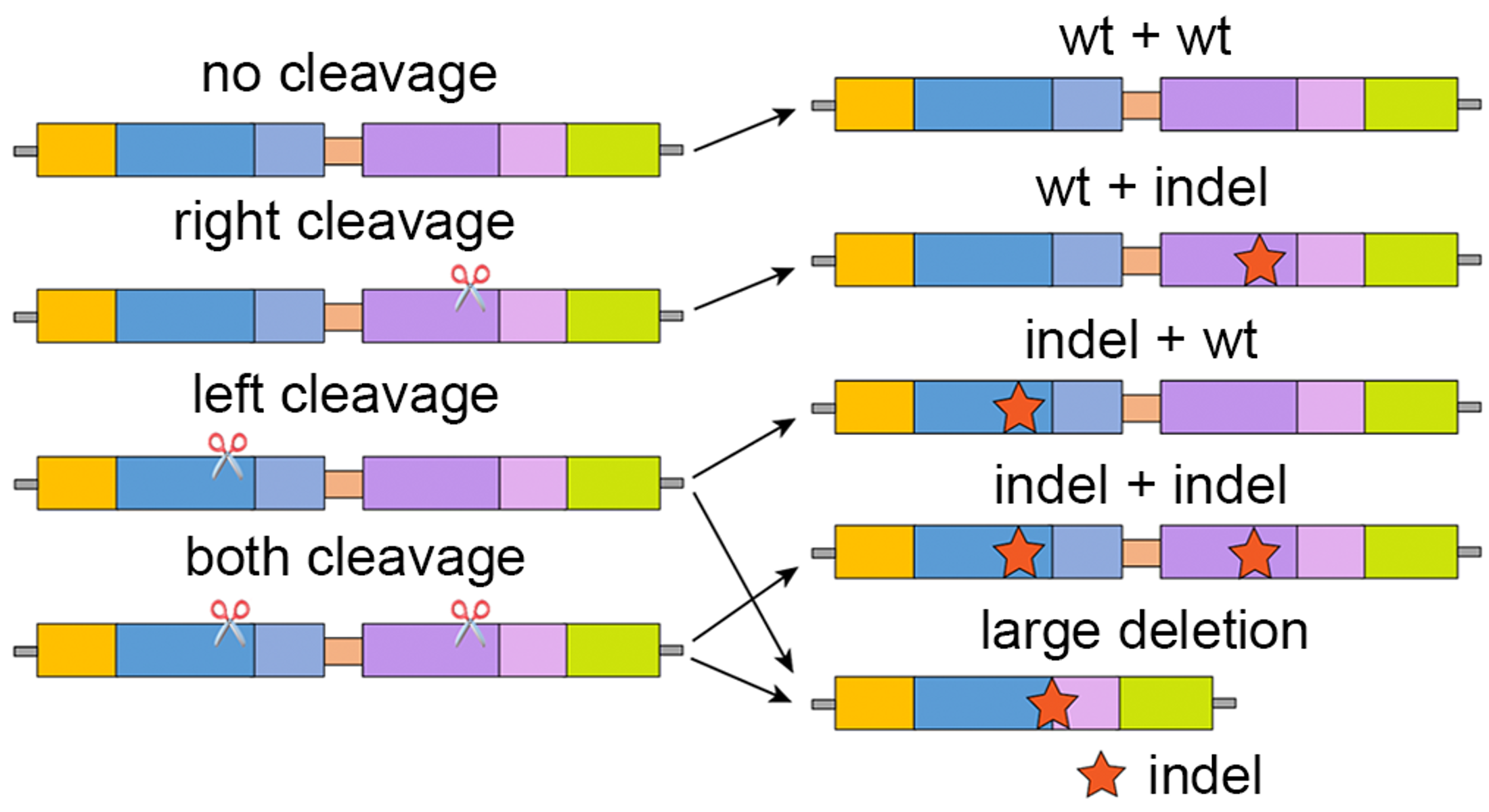
Figure 3. A demonstration of the cleavage-editing model of the dual-target system
Thereafter, we performed the high-throughput screens on three dual-target libraries including 276 control gRNAs with the same design as two tested gRNAs above, 1,902 random gRNAs with 7 off-target sequences (3 targets with 1-mismatch, 3 with 2-mismatch and 1 with 3-mismatch), and 35 benchmark gRNAs collected from previous in vitro or in vivo studies. For a head-to-head comparison of different systems, we also generated a single-target library composed of the same sets of off- and on-targets as one of the three dual-target libraries. The results showed that our dual-target system could largely reduce the experimental variations and biases, and accurately assess off-target effects on benchmark gRNAs detected both in vitro and in vivo across the genome as compared to the single-target system.
With the proven robustness of our system, we decomposed a set of sequence rules based on our large datasets, involving 2 factors in off-targeting: (1) a guide-intrinsic mismatch tolerance (GMT) independent of the mismatch context and (2) an “epistasis-like” combinatorial effect of multiple mismatches, which both are associated with the free-energy landscape in R-loop formation and explainable by a multi-state kinetic model. Then these sequence rules led to the development of MOFF, a model-based predictor of Cas9-mediated off-target effects that accounts for three components corresponding to the multiplication of individual mismatch effect (IME), the combinatorial effect (CE), and the GMT effect (Figure 4). Compared to existing methods for the in silico prediction of Cas9 off-targeting, MOFF showed a superior performance to predict the off-target effects of gRNA-target pairs as well as genome-wide gRNA specificity.
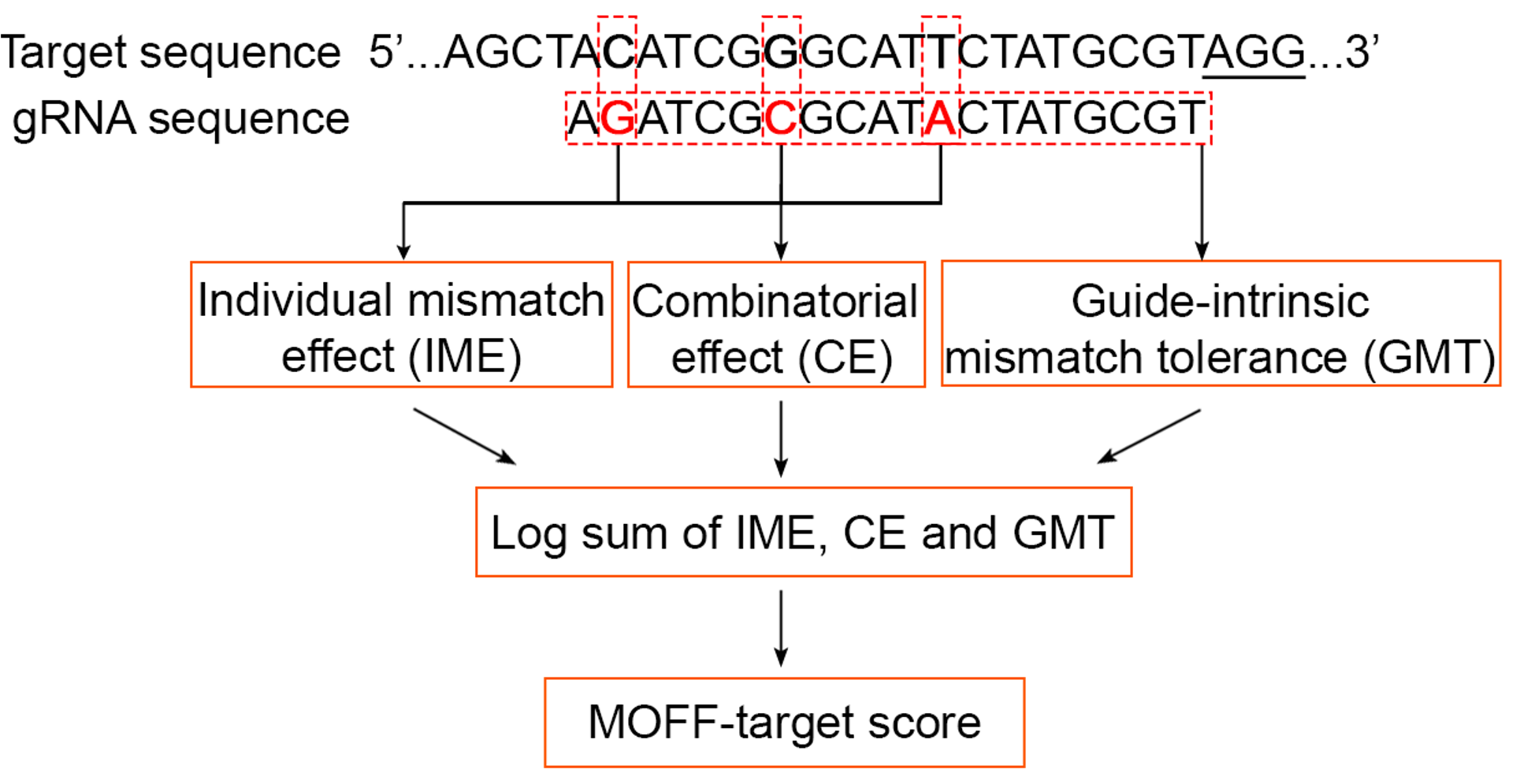
Figure 4. A schematic representation of the workflow of MOFF-target
In addition, the “epistasis-like” combinatorial effect suggested a strategy of allele-specific genome editing using mismatched guides (Figure 5). With the aid of MOFF prediction, this strategy could significantly improve the selectivity and expanded the application domain of Cas9-based allele-specific editing, as tested in a high-throughput allele-editing screen on 18 cancer hotspot mutations using the dual-target design.

Figure 5. A conceptual illustration of improving the selectivity of allele-specific editing using mismatched gRNA exemplified by KRAS G12D sequence
Our lab has developed a series of computational tools to aid in performing genetic functional screens and the analysis of protein-DNA interactions, such as recent ProTiler [8] and GuidePro [9]. If you are interested in these tools, please refer to the link (https://www.mdanderson.org/research/departments-labs-institutes/labs/xu-laboratory/resources.html) to find our resources.
References
[1] Xu, Han, et al. "Sequence determinants of improved CRISPR sgRNA design." Genome research 25.8 (2015): 1147-1157.
[2] Doench, John G., et al. "Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9." Nature biotechnology 34.2 (2016): 184-191.
[3] Allen, Felicity, et al. "Predicting the mutations generated by repair of Cas9-induced double-strand breaks." Nature biotechnology 37.1 (2019): 64-72.
[4] Shen, Max W., et al. "Predictable and precise template-free CRISPR editing of pathogenic variants." Nature 563.7733 (2018): 646-651.
[5] Chen, Wei, et al. "Massively parallel profiling and predictive modeling of the outcomes of CRISPR/Cas9-mediated double-strand break repair." Nucleic acids research 47.15 (2019): 7989-8003.
[6] Feldman, David, et al. "Lentiviral co-packaging mitigates the effects of intermolecular recombination and multiple integrations in pooled genetic screens." BioRxiv (2018): 262121.
[7] Hegde, Mudra, et al. "Uncoupling of sgRNAs from their associated barcodes during PCR amplification of combinatorial CRISPR screens." PloS one 13.5 (2018): e0197547.
[8] He, Wei, et al. "De novo identification of essential protein domains from CRISPR-Cas9 tiling-sgRNA knockout screens." Nature communications 10.1 (2019): 1-10.
[9] He, Wei, et al. "GuidePro: A multi-source ensemble predictor for prioritizing sgRNAs in CRISPR/Cas9 protein knockouts." Bioinformatics 37.1 (2021): 134-136.
Follow the Topic
-
Nature Communications
An open access, multidisciplinary journal dedicated to publishing high-quality research in all areas of the biological, health, physical, chemical and Earth sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Women's Health
Publishing Model: Hybrid
Deadline: Ongoing
Biosensing
Publishing Model: Hybrid
Deadline: Sep 30, 2026



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in