Targeting PP2A-dependent autophagy enhances sensitivity to ruxolitinib in JAK2V617F myeloproliferative neoplasms
Published in Cancer
Purpose:
The Myeloproliferative Neoplasms (MPN) are chronic hematological malignancies, described as an excessive clonal expansion of myeloid lineage cells. The prevalent expression of the JAK2V617F mutated tyrosine kinase observed in these disorders led to the development of JAK1/JAK2 inhibitors such as ruxolitinib, now in clinical use (1). Indeed, a majority of patients interrupt their treatment because of partial response or hematological side effects mainly due to a lack of specificity towards tumor cells. Therefore, there is a critical need for developing new therapeutic approaches to enhance MPN patient care. We thus hypothesized that autophagy, a catabolic process involved in cancer cell biology (2), could contribute to ruxolitinib resistance in these blood cancers. Autophagy is a dynamic process of degradation and recycling of cell compounds. It is tightly regulated and it allows cells to adapt in response to many stresses. Over the past years, extensive data have established a close relationship between autophagy and cancer. Indeed, tumor cells can hijack autophagy to their own benefits to proliferate and survive in stressful environment and even to resist cancer treatments (3). Until now, little attention has been paid to the role of autophagy in JAK2V617F MPN cells. We thus aimed to characterize the involvement of autophagy in the resistance mechanisms to ruxolitinib in these cancer types.
Results:
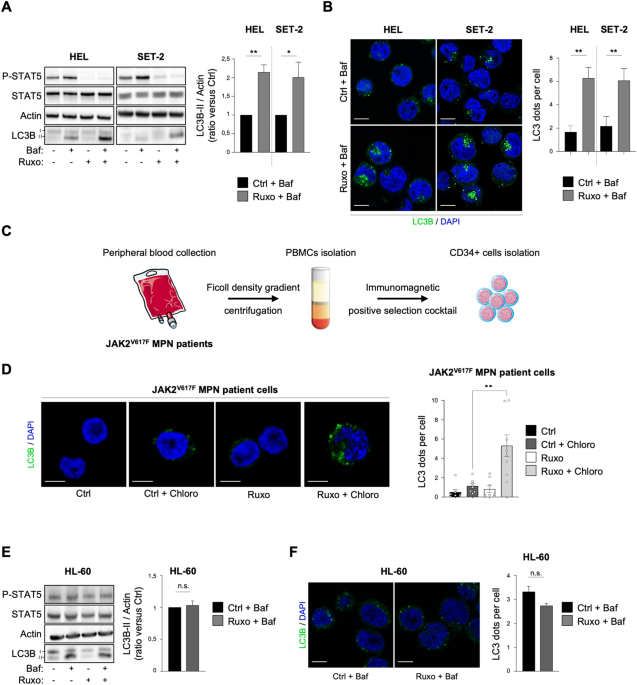
i- Ruxolitinib enhances autophagy in JAK2V617F cells.
We first measured the impact of ruxolitinib treatment on autophagy in two human cell lines and primary cultures of MPN patient samples that harbor the JAK2V617F mutation. To do so, we monitored in those cells the accumulation of the autophagosomal marker LC3B-II using both western blot and immunofluorescence experiments. We thus showed that JAK2V617F inhibitor ruxolitinib leads to an early autophagy flux induction in JAK2V617F cell lines and primary MPN patient cells. However, the autophagy flux remained unchanged in JAK2 wild-type cells upon ruxolitinib treatment. Altogether, these data demonstrate that autophagy is induced upon ruxolitinib treatment only in JAK2V617F-positive cells.
ii- Targeting autophagy increases ruxolitinib efficacy in JAK2V617F cells.
Next, we wondered whether ruxolitinib-induced autophagy could be involved in treatment outcome in JAK2V617F cells. To address this question, we treated both JAK2V617F cell lines and MPN patient samples with ruxolitinib plus different autophagy inhibitors (e.g., chloroquine, SAR405). These combinations reduced proliferation and increased death only of JAK2V617F cell lines. Accordingly, proliferation and clonogenic potential of JAK2V617F -driven primary MPN patient cells, but not of normal hematopoietic cells expressing JAK2 wild-type, were markedly impaired by the association, ruxolitinib plus autophagy inhibitors, compared to ruxolitinib alone. These results therefore indicate that ruxolitinib-induced autophagy is cytoprotective, contributes to treatment resistance of tumor cells and represents a JAK2V617F MPN cell-specific resistance mechanism.
iii- Protein-phosphatase 2A (PP2A) is involved in ruxolitinib-induced autophagy.
Mechanistically, we demonstrate that ruxolitinib-induced autophagy relies on PP2A protein phosphatase activation. Indeed, ruxolitinib treatment enhances PP2A activity in JAK2V617F cells and inhibiting this phosphatase prevents the autophagy induction mediated by JAK2 inhibition. Therefore, targeting PP2A mirrors the effects obtained with autophagy inhibitors since it sensitizes JAK2V617F cells to ruxolitinib without impacting JAK2 wild-type healthy hematopoietic cells.
iv- Targeting autophagy enhances the survival of ruxolitinib-treated mice engrafted with JAK2V617F cells.
Finally, we confirmed the cytoprotective role of autophagy in vivo in a xenograft model of JAK2 mutated cells into NSG mice. To do so, engrafted immunodeficient NSG mice with JAK2V617F cells were subjected to a daily treatment of Lys05, a potent autophagy inhibitor, in combination with ruxolitinib. In this model, the duplet therapy, i.e., autophagy inhibitor Lys05 plus ruxolitinib, reduced the tumor cells engraftment observed in the mice’s bone marrow and significantly extended mice overall survival compared with ruxolitinib alone.
Conclusion:
To conclude, autophagy or its upstream regulator PP2A represent new therapeutic targets for JAK2V617F -positive MPNs. Combining ruxolitinib with autophagy or PP2A inhibitors could enhance treatment efficacy and specificity towards the malignant clone to improve MPN patient care especially by reducing side effects. Collectively, these data pave the way for developing a clinical trial to assess the efficacy of ruxolitinib and autophagy, or its upstream regulator PP2A, inhibition in MPN patients expressing JAK2V617F.
References:
(1) Vainchenker, W., Leroy, E., Gilles, L., Marty, C., Plo, I., and Constantinescu, S.N. (2018). JAK inhibitors for the treatment of myeloproliferative neoplasms and other disorders. F1000Research 7, 82.
(2) Galluzzi, L., Pietrocola, F., Bravo-San Pedro, J.M., Amaravadi, R.K., Baehrecke, E.H., Cecconi, F., Codogno, P., Debnath, J., Gewirtz, D.A., Karantza, V., et al. (2015). Autophagy in malignant transformation and cancer progression. EMBO J. 34, 856–880.
(3) Joffre, C., Ducau, C., Poillet‐Perez, L., Courdy, C., Mansat‐ De Mas,V. (2021). Autophagy a Close Relative of AML Biology. Biology 10, 552.
Follow the Topic
-
Blood Cancer Journal

This journal seeks to publish articles of the highest quality related to hematologic malignancies and related disorders.

Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in