The apple does fall far from the tree: a new antibiotic to tackle multiresistant bacteria
Published in Chemistry
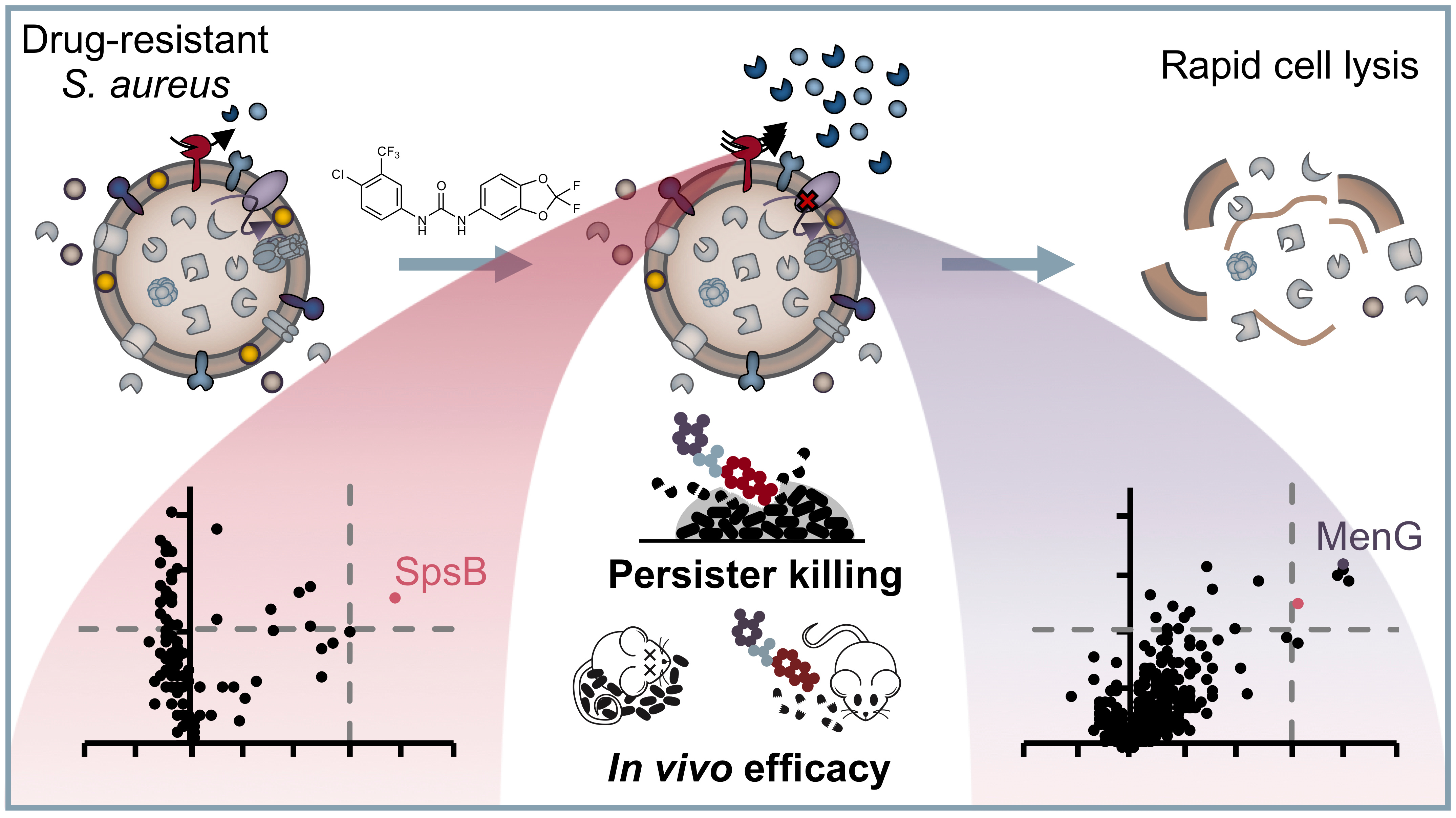
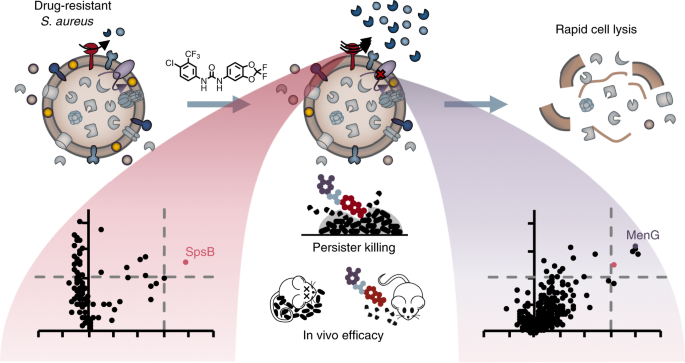
Antibiotic resistance is a worldwide growing problem and has just recently been stressed again by the report of the US Center for Disease Control and Prevention[1]. This issue is exacerbated by the fact that virtually all antibiotics in clinical use address only a small set of different targets, thereby facilitating development of cross-resistance[2]. On the contrary, other disciplines showed us that there is a class of druggable protein targets with high therapeutic potential: protein kinases. They regulate essential signalling pathways within all domains of life, in eukaryotic cells as well as in bacteria. Given the growing number of successful kinase inhibitors in cancer chemotherapy, it seems rather surprising that not a single antibacterial drug has yet been developed which acts via an analogous pathway[3]. That is why we went on a repurposing strategy to screen known kinase inhibitors for antibacterial activity against the pathogen Staphylococcus aureus. And indeed, we identified the anti-cancer drug sorafenib as major hit exhibiting antibacterial activity, which was in accordance with previous findings[4,5]. Starting from there, we were asking two questions: First, how can we further improve the chemical scaffold to increase activity and specificity against bacteria? Second, what is the compound’s mechanism of action?
For the first question, we conducted a structure-activity relationship study of 72 analogues and came up with an improved compound called PK150, which showed a ten-fold increase in antistaphylococcal activity. More importantly, PK150 was also active against methicillin-resistant S. aureus and a panel of other hard-to-treat Gram positive pathogens. PK150 proved to be efficacious against challenging persister cells and biofilms and exhibited promising activity in an in vivo infection model in mice, combined with oral bioavailability and favourable pharmacokinetic behaviour. These properties, which make PK150 a promising candidate for prospective drug development, are likely the consequence of its derivation from an already marketed drug and underline the power of drug repurposing.
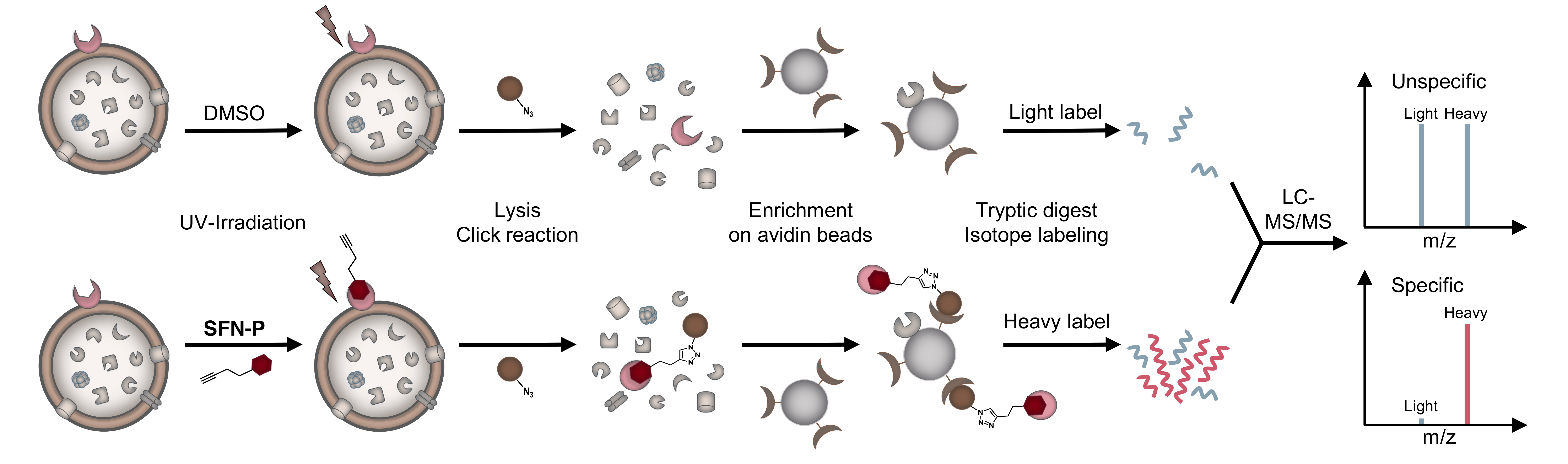
Figure 1: Schematic experimental workflow for target identification by affinity-based protein profiling (AfBPP). Intact cells are treated with probe or DMSO (as control), UV-irradiated, lysed and labelled proteins clicked to rhodamine-biotin-azide. Following enrichment on avidin beads, proteins are enzymatically digested and finally appended to either light (L) or heavy (H) isotopes via dimethyl labelling. Ratios of H/L peptides are determined via subsequent LC-MS/MS measurements.
For the second question, we equipped the compound scaffold with a photoreactive diazirine moiety and an alkyne tag to perform affinity-based protein profiling (Figure 1)[6,7]. This technique allows to create a covalent cross-link between the chemical probe and the target protein and to subsequently enrich and identify these proteins by LC-MS/MS measurements. By doing so, we identified two completely different targets, which we validated with different experiments. To our surprise, none of the identified proteins were actually kinases. Instead, the compounds were shown to inhibit menaquinone synthesis, which is required for energy metabolism, and to stimulate protein secretion by over-activation of signal peptidase Ib (SpsB). Particularly the latter finding is intriguing, as protein activation instead of inhibition is a rare event in small molecule-protein interactions and has also not been reported for this enzyme yet. However, several independent experiments confirm this observation and led us to the hypothesis that it might be a resulting over-abundance of autolytic enzymes that might cause cell wall degradation and finally cell lysis. We are currently looking deeper into the molecular mechanisms that drive SpsB over-activation and hope that this might aid in even improved compound development and a better understanding of how this important bacterial enzyme works.
Taken together, what started with the idea to target bacterial kinases ended in a completely different direction. It’s one of these examples that demonstrate that you can never predict where science will lead you. Though, what we kept was our goal to uncover new pathways to address antimicrobial resistance, and despite all the difficulties that still lie ahead of us to really end up with a clinical drug, we think this work demonstrates that there are plenty of possibilities out there to tackle bacteria in unprecedented ways – they just need to be discovered.
For more information, you can find our paper here.
References:
[1] CDC. Antibiotic resistance threats in the United States, 2019. Atlanta, GA: U.S. Department of Health and Human Services, CDC; 2019.
[2] M. F. Chellat, L. Raguž, R. Riedl: Targeting Antibiotic Resistance, Angew. Chem. Int. Ed. 55, 2–30 (2016).
[3] Kurosu, M. & Begari, E. Bacterial protein kinase inhibitors. Drug Dev. Res 71, 168–187 (2010).
[4] Chang, H.-C. et al. In vitro and in vivo activity of a novel sorafenib derivative SC5005 against MRSA. J. Antimicrob. Chemother. 71, 449–459 (2016).
[5] Roberts, J. L. et al. GRP78/DNA K is a target for Nexavar/Stivarga/Votrient in the treatment of human malignancies, viral infections and bacterial diseases. J. Cell. Physiol. 230, 2552–2578 (2015).
[6] Evans, M. J. & Cravatt, B. F. Mechanism-based profiling of enzyme families. Chem. Rev. 106, 3279–3301 (2006).
[7] Fonović, M. & Bogyo, M. Activity-based probes as a tool for functional proteomic analysis of proteases. Expert Rev. Proteomics 5, 721–730 (2008).
Follow the Topic
-
Nature Chemistry
A monthly journal dedicated to publishing high-quality papers that describe the most significant and cutting-edge research in all areas of chemistry, reflecting the traditional core subjects of analytical, inorganic, organic and physical chemistry.



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in