The first covalent inhibitor for aminoacyl-tRNA synthetases
Published in Microbiology

AARSs are underexploited therapeutic targets
Aminoacyl-tRNA synthetases (AARSs) are housekeeping enzymes catalyzing the formation of an ester bond between a specific amino acid and its cognate tRNA. They undertake the essential function of protein synthesis in all cells, including pathogenic microorganisms. Despite their similarity across organisms, sequence and topological differences between pathogenic microbial AARSs and human AARSs make it possible to design drugs that selectively inhibit pathogen AARSs. AARS inhibitors have yet been developed into drugs, including mupirocin, approved for the treatment of skin infections, halofuginone, approved as a veterinary drug for treating coccidiosis, also received orphan drug designation for the treatment of systemic sclerosis, and AN2690, approved for the treatment of onychomycosis. More AARS inhibitors are being actively developed. Different types of AARS inhibitors have been found, including substrate mimetics, Trojan horses, induced-fit inhibitors, and reaction hijacking inhibitors. However, covalent inhibitors of AARS have not been reported.
Obafluorin (OB) forms a covalent bond with Tyr462 of ThrRS
Threonyl-tRNA synthetase (ThrRS) inhibitors such as borrelidin have been shown to have a wide range of biological activities, including antimicrobial, antimalarial, and antiangiogenic activities. Recently, a novel natural product, obafluorin (OB), produced by Pseudomonas fluorescens ATCC 390502, was identified as a potent inhibitor of bacterial ThrRS. OB contained a β-lactone ring with a 2,3-dihydroxybenzamidyl moiety attached to the α position and a 4-nitrobenzyl moiety attached to the β position. It showed no apparent structural similarities with the three substrates of ThrRS: L-threonine, ATP, and tRNA.
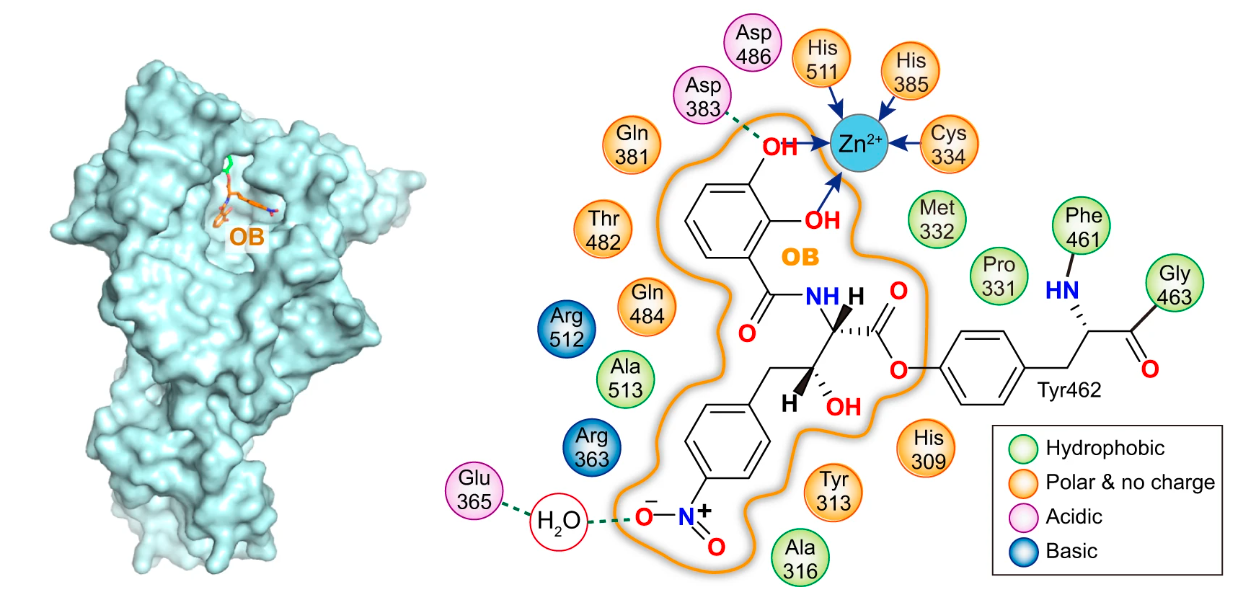
To elucidate the mechanism by which OB inhibits ThrRS, we cocrystallized a fragment of Escherichia coli ThrRS containing catalytic and anticodon-binding domains (residues 242–642) with OB and determined the structure to a resolution of 2.5 Å. In the two catalytic domains of the ThrRS homodimer, OB was bound to the active site for Thr-AMP formation. Interestingly, the four-membered ring of the β-lactone of OB was opened in the ThrRS–OB structure. Instead, the acyl group formed a new ester bond with the phenolic group of the Tyr462. In addition, the two hydroxyl groups on the o-diphenol moiety of OB formed two coordination bonds with the conserved catalytic Zn2+ ion of ThrRS. This structure suggested that OB inhibited ThrRS by forming coordination bonds with the Zn2+ ion in the catalytic center of ThrRS, allowing its β-lactone ring to be approached and attacked by the phenolic group of Tyr462, and finally forming a new covalent bound with Tyr462.

Fig. 1. OB forms a covalent bond with the Tyr462 residue of ThrRS.
OB covalently modifies engineered lysine and arginine
We hypothesized that if the Tyr462 was mutated to a nucleophilic residue in similar side chain length, the mutant ThrRS should also be covalently inhibited by OB. To test this hypothesis, we purified the ThrRS_Y462K and ThrRS_Y462R mutant proteins, and developed a thermal shift assay (TSA) which could rapidly evaluate the effect of the inhibitor. In the TSA experiment, OB increased the Tm of ThrRS_WT by 34.8 °C. Interestingly, OB also increased the Tm of ThrRS_Y462K by 36.8 °C, suggesting that OB was able to form a covalent bond with the engineered Lys462. Not surprisingly, OB only increased the Tm of ThrRS_Y462R by 6.5 °C, since arginine was a weak nucleophilic residue. The solved crystal structure of both the ThrRS_Y462K mutant and ThrRS_Y462R mutant were very similar to that of the wild-type protein with OB in the catalytic pocket, suggesting that lysine and arginine also reacted with β-lactone ring of OB to form a covalent bond.
OB prevents ThrRS from binding to all three substrates
The ThrRS–OB structure showed that the o-diphenol group of OB occupied the position of l-threonine and replaced it to bind Zn2+ ion and Asp383. At the same time, the conformations of Gln484 and Met332 were changed to accommodate OB. On the other hand, the opening of the β-lactone ring made OB exhibit a more extended molecular configuration. This allowed its nitrobenzene group to extend to the other side of the pocket and form stacking interactions with Tyr313 and Arg363, two important residues for ThrRS to bind the 3' end of tRNA. Therefore, the nitrobenzene group of OB occupied the binding site of tRNA A76, which prevented tRNA from entering the active center of ThrRS.
To test the mutually exclusive effect between OB and ATP, we engineered a ThrRS_Y462F mutant that mutated Tyr462 to a phenylalanine to avoid covalent bonding with OB. As a result, in the TSA experiment, OB only increased the Tm of ThrRS_Y462F by 2.6 °C. Consistently, OB failed to inhibit the activity of ThrRS_Y462F in the ATP hydrolysis assay up to 5 μM. More interestingly, when we cocrystallized ThrRS_Y462F with OB and ATP, ATP was visible in the active site of ThrRS together with two co-bound Mg2+ ions. No densities for OB were observed in the active center. Without the binding of OB, the conformation of Tyr313 of ThrRS_Y462F was in a position that would clash with OB as it was in the ThrRS–OB complexes. In addition, compared with the ThrRS_Y462F–ATP structure, OB induced a significant conformational change to the outer side of the active cleft, which was similar to that induced by another potent ThrRS inhibitor, borrelidin. Therefore, OB prevents ThrRS from binding to all three substrates, including l-threonine, tRNA, and ATP.
Discussion
Advances in covalent drug discovery have led to successful drugs, including inhibitors of epidermal growth factor receptor (EGFR), Bruton’s tyrosine kinase (BTK), KRAS(G12C), and SARS-CoV-2 main protease. The discovery that OB can form a covalent bond with Tyr462 of ThrRS provides a new warhead design strategy for the development of covalent inhibitors targeting tyrosine. This work not only reports OB as the first AARS covalent inhibitor, but also shows that if placed in a suitable position, β-lactone could covalently modify tyrosine, lysine, and even the weakly nucleophilic residue arginine, which are not commonly used to develop covalent inhibitors, in addition to previously reported cysteine, serine, and threonine.
References
1. Qiao, H., Xia, M., Cheng, Y. et al. Tyrosine-targeted covalent inhibition of a tRNA synthetase aided by zinc ion. Commun Biol (2023) 6, 107.
2. Kwon, N. H., Fox, P. L. & Kim, S. Aminoacyl-tRNA synthetases as therapeutic targets Nat Rev Drug Discov (2019) 18, 629-650.
3. Wells, J. S., Trejo, W. H., Principe, P. A. & Sykes, R. B.Obafluorin, a novel beta-lactone produced by Pseudomonas fluorescens. Taxonomy, fermentation and biological properties J Antibiot (Tokyo) (1984) 37, 802-803.
4. Scott, T. A. et al.Immunity-Guided Identification of Threonyl-tRNA Synthetase as the Molecular Target of Obafluorin, a beta-Lactone Antibiotic ACS Chem Biol (2019) 14, 2663-2671.
5. Fang, P. et al.Structural basis for full-spectrum inhibition of translational functions on a tRNA synthetase Nat Commun (2015) 6, 6402.
6. Boike, L., Henning, N. J. & Nomura, D. K.Advances in covalent drug discovery Nat Rev Drug Discov (2022).
Follow the Topic
-
Communications Biology
An open access journal from Nature Portfolio publishing high-quality research, reviews and commentary in all areas of the biological sciences, representing significant advances and bringing new biological insight to a specialized area of research.

Related Collections
With Collections, you can get published faster and increase your visibility.
Artificial Intelligence Methodology in Structural Biology
Publishing Model: Hybrid
Deadline: Nov 30, 2026
Healthy Aging
Publishing Model: Open Access
Deadline: Dec 31, 2026

Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in