The Peter Pan syndrome: stalled development in pediatric brain tumors
Published in Cancer

Over the last decades, researchers have been peering into the genomes of cancer cells to bring murky tumor entities into sharp focus… and revealed an enigma. Pediatric brain tumors can be divided into distinct molecular subtypes, each characterized by few genetic alterations that, more often than not, involve developmental pathways. Curiously, these entities have recurrent spatial and temporal patterns, with each tumor type arising in a distinct age window and brain region. What makes some cells and not others vulnerable to oncogenic alterations? Understanding the origins of these tumors has been a quest in our labs and others for many years.
The catch: it is difficult to pinpoint the cell of origin of a tumor when, in many cases, we don’t even know all the cell types that are present normally during development. Defining this diversity is a daunting task, since the brain is an incredibly complex organ, with thousands of distinct cell types that vary by brain region and age. But it is the perfect moment to ask. We now have powerful, high-throughput technologies for profiling gene expression in individual cells, without targeting certain cell types a priori – or even knowing they exist.
Armed with these tools, we set out to create an atlas of cell types present during development in the brain regions where several childhood brain tumors occur. We profiled gene expression in 65,000 cells from the developing mouse forebrain and pons, and the human fetal brainstem.
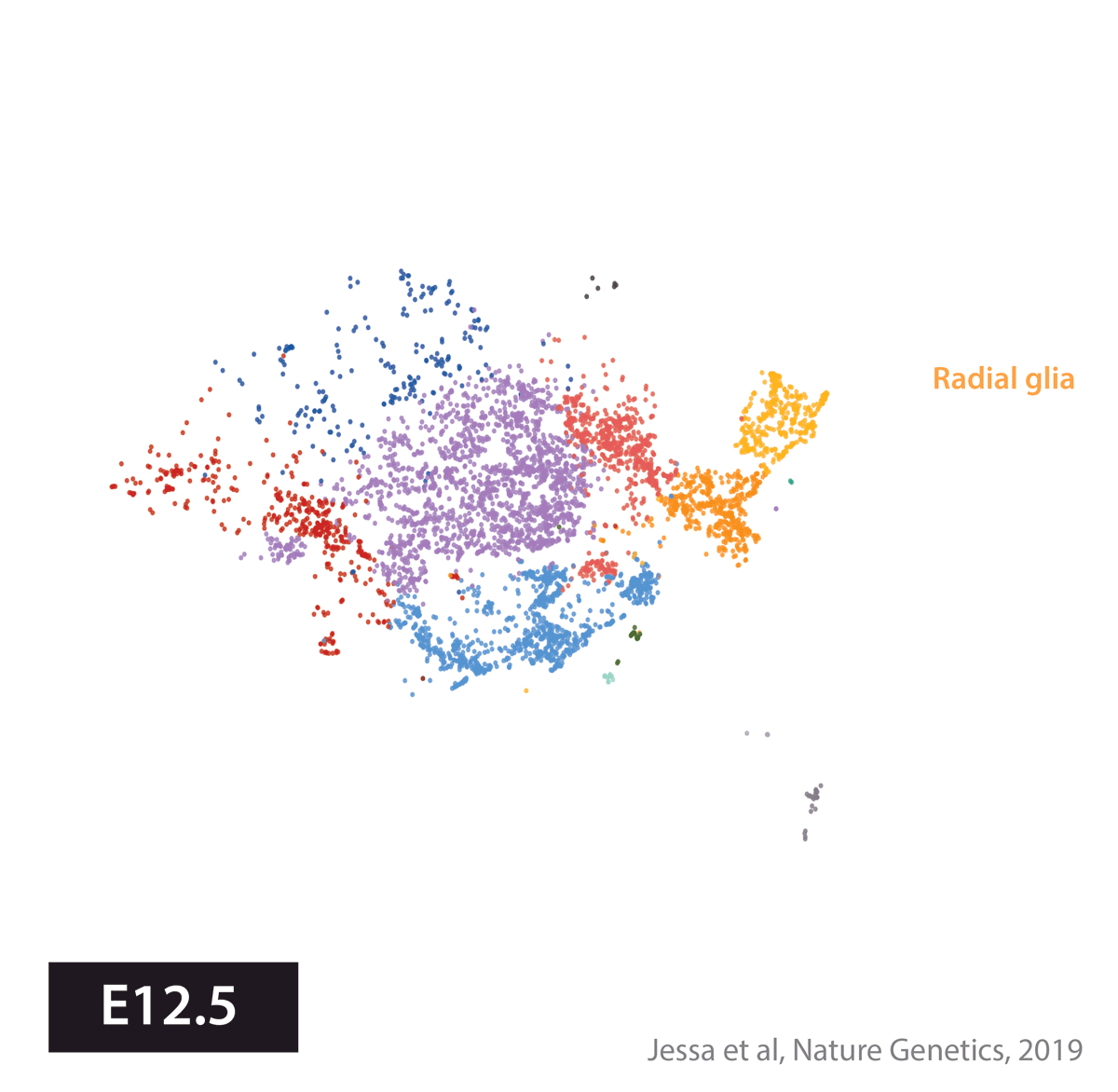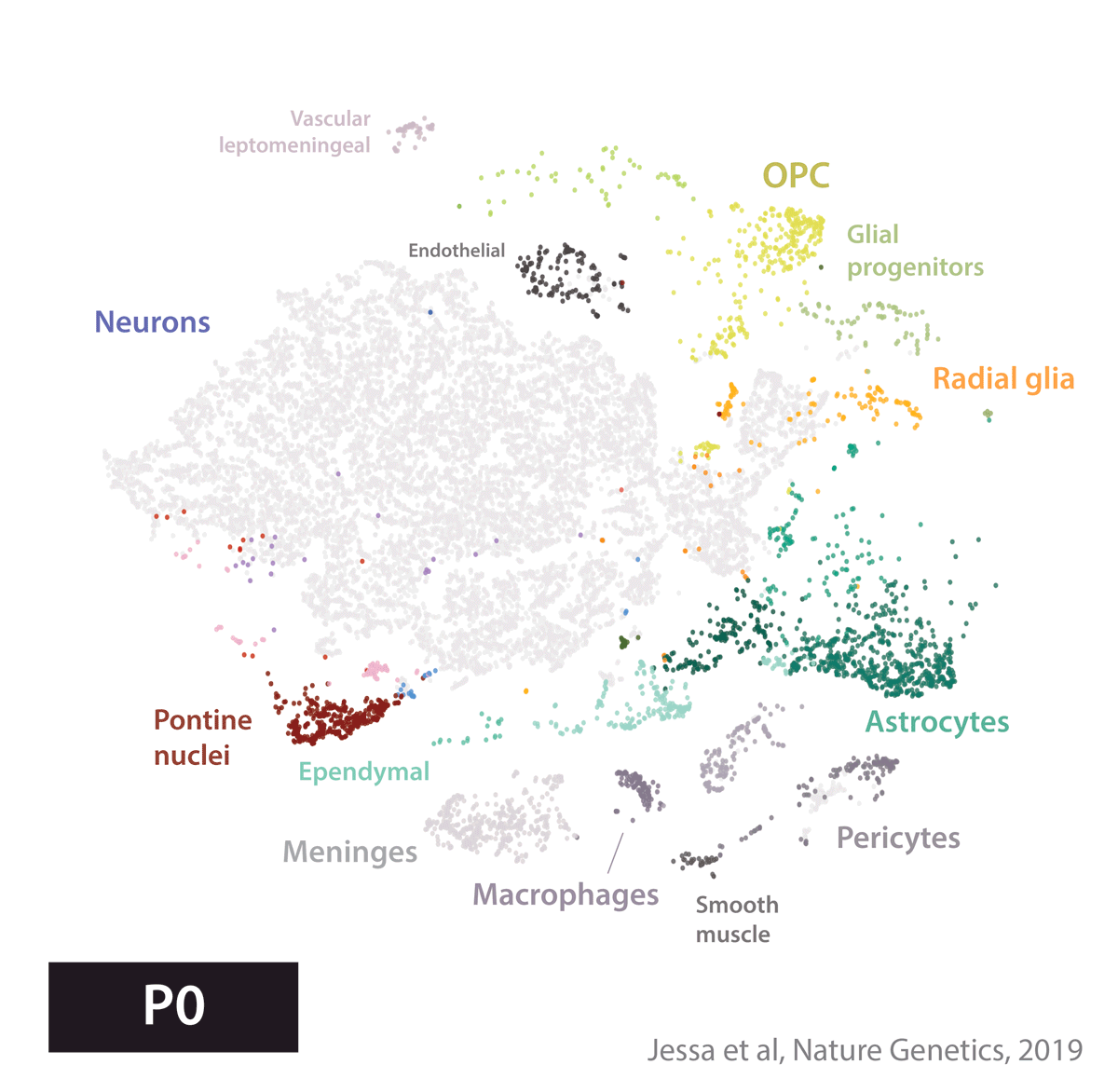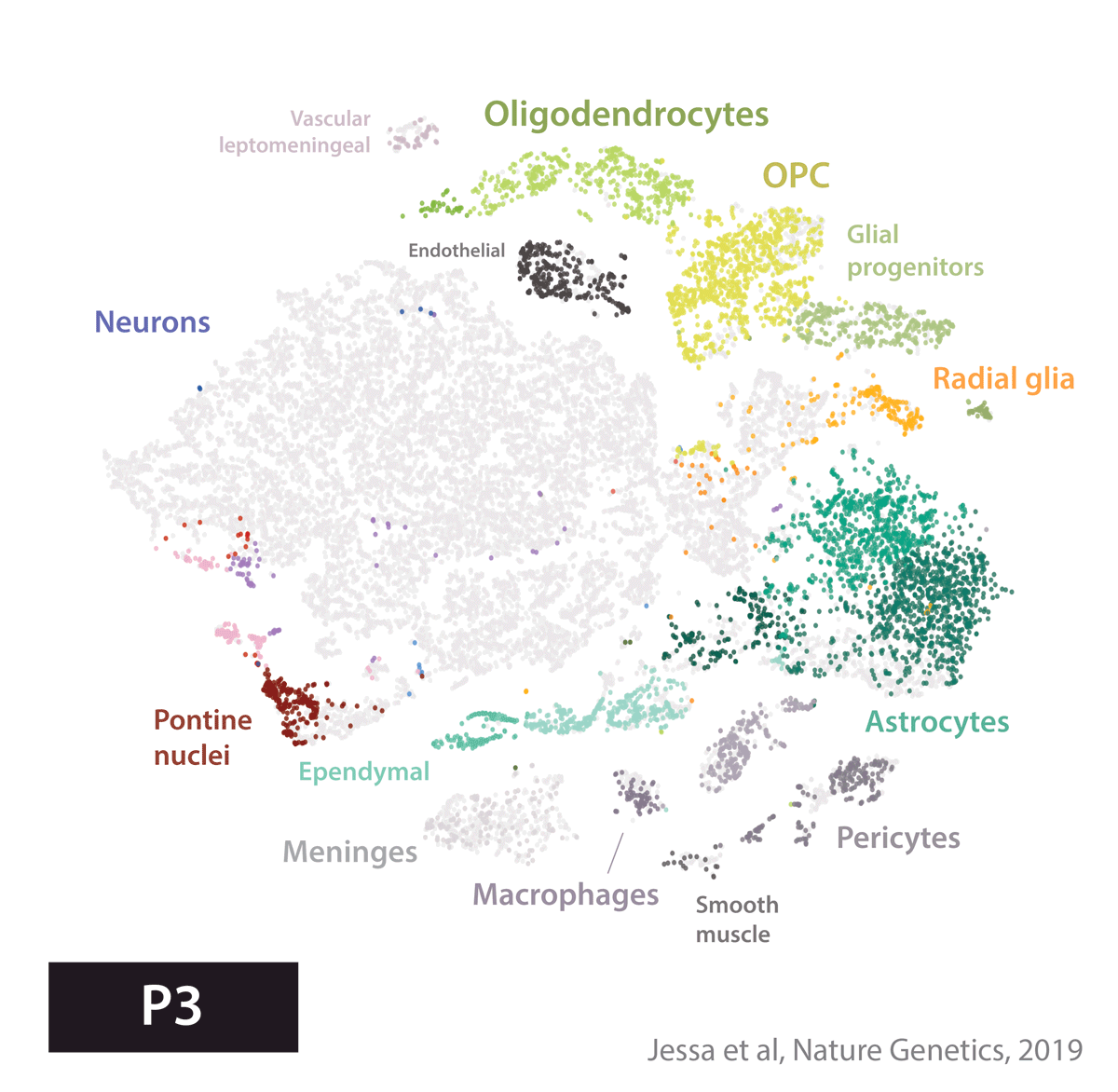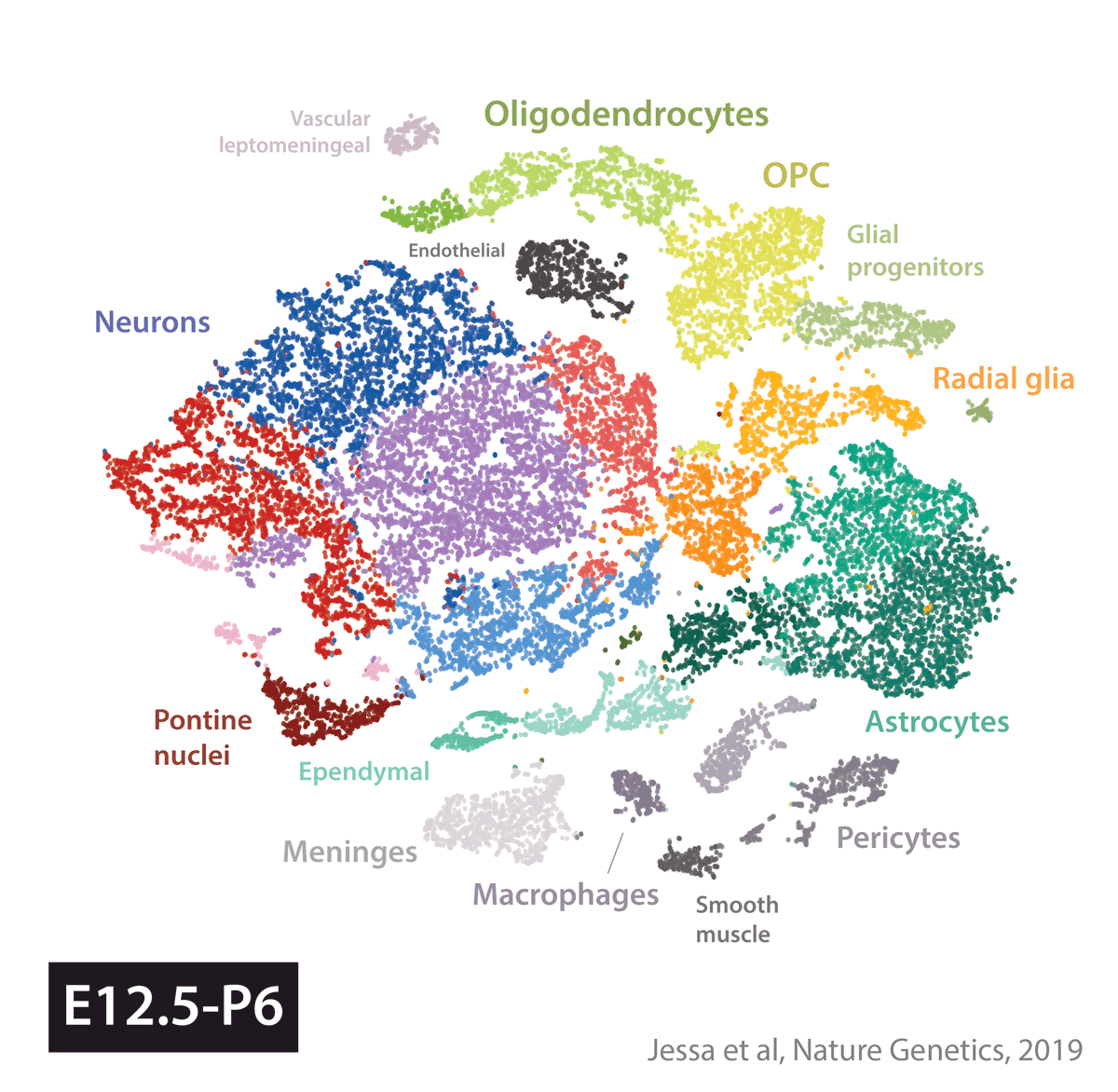
We next used this atlas as a reference, to match tumors to their most similar developmental state. In other words, we used a data-driven approach to identify cell of origin in tumors, by analyzing the cell types present in the healthy brain. Four tumor types told four very different stories.
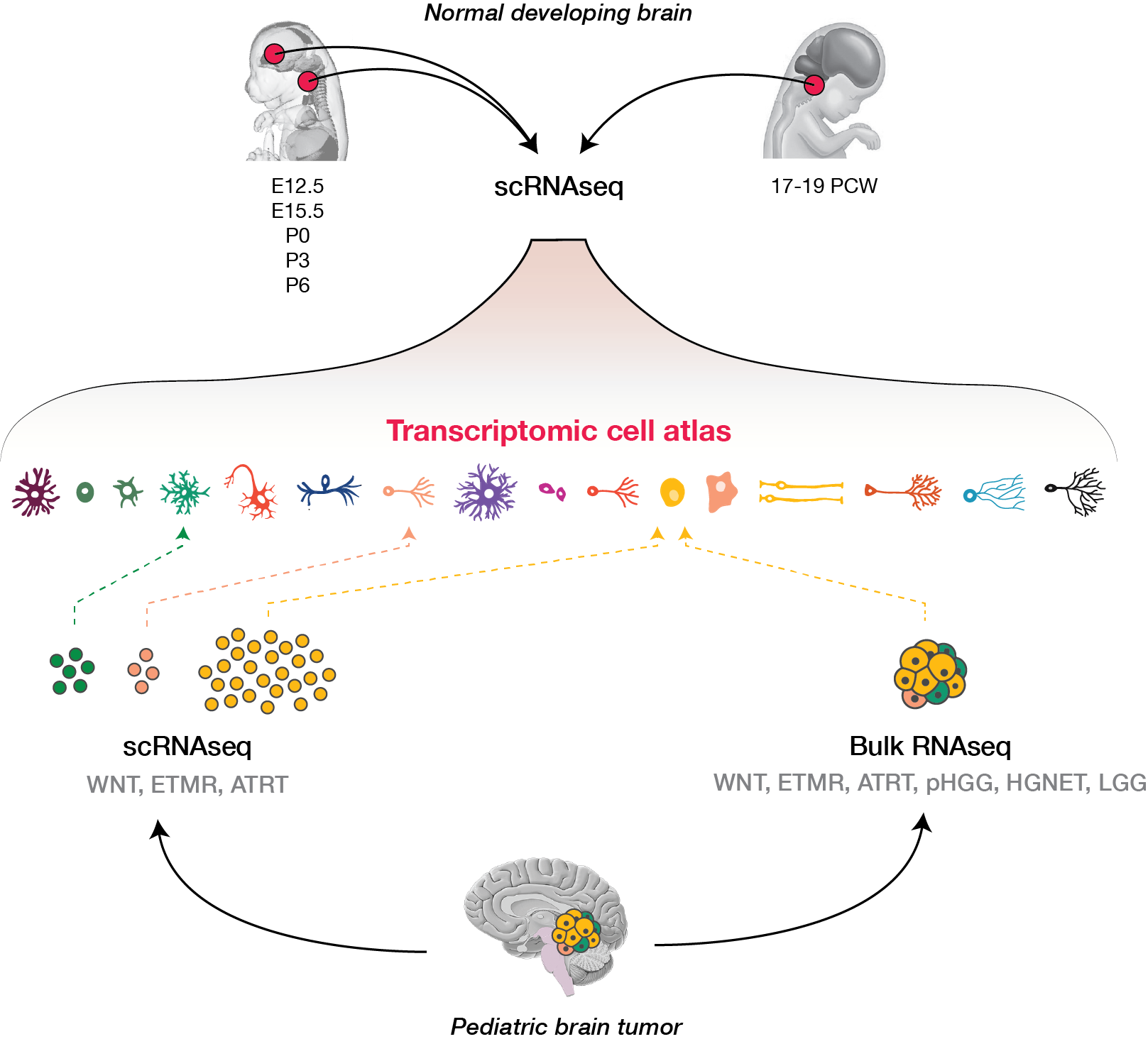
In the WNT subtype of medulloblastoma, previous work had elegantly shown that tumors likely arise from the lower rhombic lip (LRL), a progenitor domain of the hindbrain which, in mouse, is transiently present during embryonal development. This domain can give rise to several cell lineages, but mapping to our developmental atlas resolved the uncertainty and pointed to one specific cell type at the origin of these tumors, the mossy fiber neurons. A the single-cell level, WNT tumor cells resembled immature neurons.
In embryonal tumors with multilayered rosettes (ETMRs), expression of the oncogenic microRNA cluster C19MC is driven by a fusion with the TTYH1 gene. Mechanistically, to drive C19MC expression, TTYH1 would need to be normally expressed in the tumor cell of origin. Where is TTYH1 expressed? We now have the resolution to answer that question. We found that at prenatal timepoints, TTYH1 was specifically expressed in radial glial cells, the stem cells of the brain. TTYH1 expression then switched postnatally mainly to astrocytes. Since ETMRs are neuronal (not astrocytic) tumors, this nominates prenatal radial glia as candidate cells of origin of this entity. By single-cell analysis, we showed that tumors were composed of a radial glia-like population, but also more differentiated neuron-like cells.
Atypical teratoid-rhaboid tumors (ATRTs) are an interesting entity, because molecularly similar tumor types arise throughout the central nervous system as well as in soft tissues like muscle and kidney. Unlike the other tumor types, ATRTs did not match any specific cell type in our atlas. This suggests that these tumors arise from an earlier progenitor not captured in our atlas, or outside the neuro-ectoderm, and the origins of these tumors thus remain puzzling.
Finally, high-grade gliomas bearing the Histone 3 K27M mutation are thought to arise from a glial progenitor in the brain. If the K27M mutation impairs differentiation, what does a glioma cell become, when the driver alteration is removed? To answer this question, we removed the driver mutation using CRISPR/Cas9, and cultured cells in differentiation conditions. Cells without the K27M mutation progressed further along astrocytic differentiation than mutant lines. This supports the glial progenitors as cells of origin, and provides proof-of-concept that the differentiation block in these tumors may be reversible.
Altogether, through a major collaborative effort, we refined cell of origin in three pediatric brain tumor entities: WNT medulloblastomas, ETMRs, and K27M gliomas of the pons. Our data point to a common mechanism at the origin of these tumors: impaired differentiation. Like Peter Pan stuck in Never Land, specific neural progenitors acquire specific genetic alterations and become stalled in development, ultimately leading to oncogenesis.
Written by Selin Jessa, Nada Jabado and Claudia Kleinman
Follow the Topic
-
Nature Genetics
This journal publishes the very highest quality research in genetics, encompassing genetic and functional genomic studies on human and plant traits and on other model organisms.

Related Collections
With Collections, you can get published faster and increase your visibility.
Mechanisms and impact of 3D genome organisation
Publishing Model: Hybrid
Deadline: Nov 30, 2026
Human Admixture and Migration
Publishing Model: Hybrid
Deadline: Feb 28, 2027


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in