UV photochemistry of the L-cystine disulfide bridge in aqueous solution investigated by femtosecond X-ray absorption spectroscopy
Published in Chemistry, Physics, and Cell & Molecular Biology

Sulfur plays an essential role in life, in particular disulfide bonds between two cysteine amino acids in proteins provide structural stability and help regulate biological processes in the cell. Despite the importance and ubiquity of disulfide bonds, they are quite susceptible to damage and can easily be broken by, for example, ultraviolet (UV) light. In biology, such a bond cleavage contributes to a loss of protein function and in some cases diseases. However, UV-initiated disulfide bond breaking has also been harnessed for applications in chemical catalysis, sensors, pharmaceuticals, polymers and nanomaterials. For a long time, there has been some debate whether disulfides break symmetrically, resulting in two identical sulfur-centered radicals, or whether they can also break asymmetrically, yielding one sulfur-sulfur centered radical and one carbon centered radical. One influencing factor is the energy of the exciting light, with a threshold-wavelength of about 200 nm. UV light of wavelengths shorter than 200 nm can break sulfur-sulfur as well as carbon-sulfur bonds. For UV light of longer wavelengths, i.e. lower energy, the case was not clear because of a divergence between observations made on UV-excited disulfides in the gas-phase (isolated molecules) and in solution (molecules densely surrounded by solvent molecules such as water). In the gas phase, only the sulfur-sulfur bond of disulfides is broken. But in solution several accounts of carbon-sulfur bond cleavage with wavelengths above 200 nm exist.
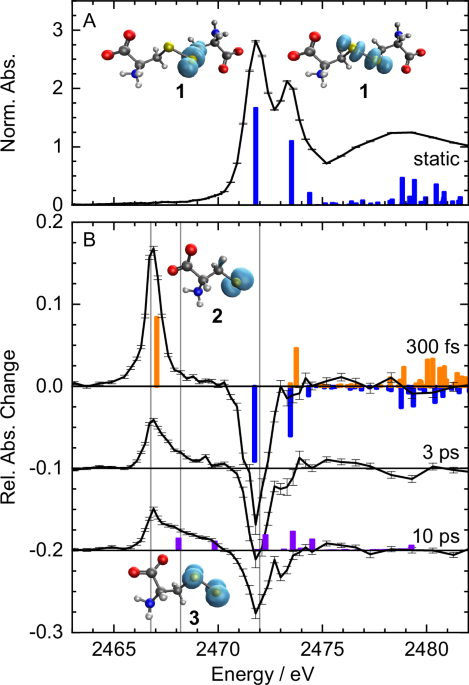
To better understand the reasons for this discrepancy between an isolated molecule and a molecule in a liquid, we investigated the photochemistry of L-cystine, the disulfide of the amino acid L-cysteine, in aqueous solution using time-resolved X-ray absorption spectroscopy (TRXAS). TRXAS is a valuable tool due to its element specificity (we only see what is happening around sulfur atoms) and its sensitivity to the chemical environment of the sulfur atoms. One of the difficulties in understanding this process is that it occurs extremely quickly - within a tenth of a millionth of a millionth of a second or 100 femtoseconds (that’s 12 zeros after the decimal point). In our experiment, the reaction is initiated by exciting the disulfide with a UV laser pulse that is 100 femtoseconds long. We are only using 0.0002 W of average UV laser power but such a short flash amounts to 60 MW of power in the light focus with which you could power 10 million LED light bulbs - admittedly, only for 100 fs. After a chosen time delay an even shorter X-ray pulse follows the UV flash to probe how the chemical reaction evolves by measuring the change in absorption of the sulfur atoms. This kind of measurement can only be done with an X-ray free-electron laser (XFEL) such as PAL-XFEL at Pohang Accelerator Laboratory in South Korea because no other type of light source can produce such short X-ray flashes.
By comparing the X-ray absorption spectra of the sulfur atoms in the chemical products with quantum chemical calculations, we were able to identify the earliest photoproducts of L-cystine in aqueous solution and follow their temporal evolution, from one hundred femtoseconds to just under a nanosecond: Within 140 fs the disulfide molecule splits symmetrically in half by rupture of the disulfide bond. This means that disulfides in gas phase and liquid phase behave initially the same. But we saw this first photoproduct vanish surprisingly fast while the parent molecule L-cystine reformed: Within 2 picoseconds (2 millionths of a millionth of second) 70 % of the disulfide bonds appeared to have reformed. This reverse reaction is impossible in the gas phase, but not uncommon in solution, where the surrounding medium keeps the reaction partners close for a while. Furthermore, we observe a second reaction product that emerged when the first product started to recombine to L-cystine. This recombination process happens at relatively high energy, that is, part of the UV light energy, which was initially absorbed, is still stored in the reformed L-cystine molecules, enough to split the carbon-sulfur bond and produce secondary products.
These results provide an answer to the old question, whether only the sulfur-sulfur bond cleavage can happen in solution or whether the carbon-sulfur bond can be broken, too. Our results clearly show the same primary reaction step as observed in isolated molecules but they also reveal the delayed emergence of other molecular sulfur species in solution which were not directly generated by the UV flash of light. This sequence of products can only be resolved with femtosecond techniques. The ultrafast and dominant reformation of the initial disulfide structure suggests that nature may use disulfide bond cleavage as a way to dynamically dissipate the destructive energy of UV light by sacrificing disulfide bonds for a short stretch of time, a mechanism that deserves further investigation in proteins.
Follow the Topic
-
Nature Communications
An open access, multidisciplinary journal dedicated to publishing high-quality research in all areas of the biological, health, physical, chemical and Earth sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Women's Health
Publishing Model: Hybrid
Deadline: Ongoing
Tumor Microenvironment Crosstalk and Therapeutic Implications
Publishing Model: Hybrid
Deadline: Nov 02, 2026




Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in