Viral infection by exosome mimicry: the HAVCR1/NPC1 pathway
Published in Microbiology
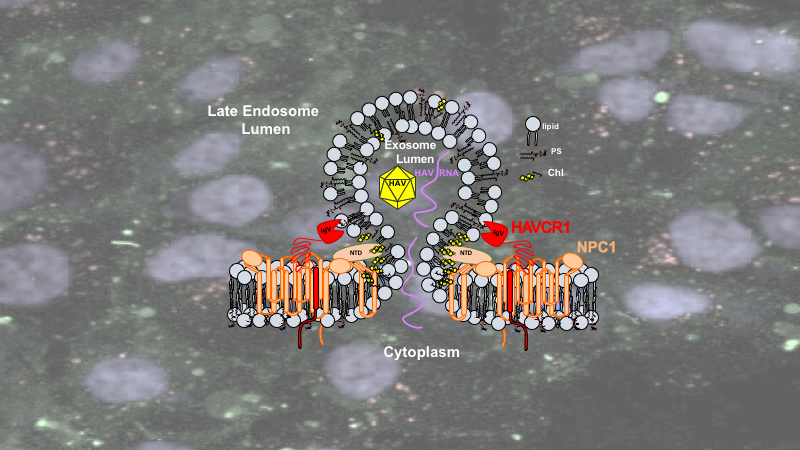

To study the exosome cargo delivery mechanism, we used exosomes from hepatitis A virus (HAV) infected cells (exo-HAV) to track cargo delivery of HAV genomes into the cytoplasm. Exo-HAV, which contains capsid-free HAV RNA and viral particles within the exosome lumen, is an ideal reagent to study exosome cargo delivery of RNA because it induces a productive HAV infection that can be easily assessed and quantitated. Using CRISPR/cas9 technology to knockout genes, we found that two lipid receptors, HAVCR1 that binds phosphatidylserine (PS) and NPC1 that binds cholesterol (Chl), are required for membrane fusion at the late endosomes (LE). We showed that this fusion process mediates cargo delivery of capsid-free RNA from the lumen of the exosomes into the cytoplasm, which avoids exosome and cargo degradation by lysosomal lipases and nucleases and triggers HAV replication. Our results indicated that the capsid-free HAV RNA contained in the lumen of the exosomes rather than the uncoating of the HAV particles within the endosome is mainly responsible for exo-HAV infection. This is consistent with the partial sensitivity of HAV to pathogen reduction technologies based on photochemical reactions to inactivate pathogens in blood and blood products, which use membrane-permeant compounds that inactivate capsid-free HAV RNA but cannot penetrate the tight viral capsid to inactivate the HAV genomic RNA. This envelope-glycoprotein-independent exosome cargo delivery pathway is mediated by two lipid receptors that are anchored to the LE membrane by their transmembrane domains and bound to the exosome by their lipid-binding domains, which form bridges that bring the LE and exosome membranes to close proximity. Our results favor a model in which the contact of the LE and exosome delimiting membranes induces hemifusion that progresses to complete fusion in a Chl-rich environment to deliver the cargo into the cytoplasm (Fig. 1, left panel). Previous work showed that Ebola virus (EBOV) usurps this same HAVCR1/NPC1 pathway to deliver its genetic material into the cytoplasm and initiate infection. However, EBOV has an envelope glycoprotein (GP) that is believed to mediate fusion of the viral envelope with the delimiting membrane of the LE (Fig. 1, middle panel). This exosome fusion mechanism, which is mediated by HAVCR1 and NPC1 at the low pH of the LE in the absence of a viral envelope glycoprotein, provides a new understanding of the exosome cargo delivery process and how viruses use exosome mimicry to infect cells. However, it raises questions about the role of viral envelope glycoproteins in the LE fusion process. Perhaps these viral glycoproteins stabilize or increase the size of the fusion pores to allow the passage of the viral nucleoproteins into the cytoplasm. Our findings offer new therapeutic targets to block virus infection and a framework to reassess the role of envelope glycoproteins in virus cell entry. Further research is needed to fully understand the exosome cargo delivery and viral exosome mimicry mechanisms, which will lead to the identification of additional cellular factors involved in these processes.
Our work further supports the notion that exo-HAV is a bona fide exosome capable of delivering HAV genomes into the cytoplasm to begin HAV replication. Indeed, exo-HAV is not a “quasi-enveloped HAV (eHAV)” as suggested by others1 but an exosome that fuses at the LE and delivers an RNA cargo from the exosome lumen into the cytoplasm, as described for many other exosomes from infected or uninfected cells. The ability of viruses to infect cells independently of envelope-receptor interactions was described two-decades ago as the Trojan exosome hypothesis2 and shown for many enveloped viruses including HIV and HCV3. This hypothesis can now be extended to non-enveloped viruses based on our results showing that luminal capsid-free HAV RNA is mainly responsible for the infectivity of exo-HAV. It is likely that the HAV particles observed in the lysosome4,5 represent a late stage in the degradation process of endocytosed macromolecules that do not lead to productive infection.
In our previous work, we showed that HAVCR1 is an HAV cellular receptor6-10, and that soluble forms of HAVCR1 neutralize HAV particles11-13 inducing conformational changes that alter the viral particles and release the viral genome12. Because the HAV capsid is very stable at low-pH and high-temperature, a receptor-mediated alteration of the viral particles is required to deliver the HAV genome into the cytoplasm for HAV replication (Fig. 1, right panel). We and others showed that HAVCR1 is a PS receptor that binds apoptotic cells14-16 and, in our current work, exosomes produced in HAV-infected cells. HAV, like many other viruses, has multiple cellular receptors9,10. It has recently been suggested that Integrin β14 and gangliosides5 are involved in the cell entry of HAV particles and exo-HAV. However, these two molecules do not induce conformational changes in HAV particles leading to the uncoating of the HAV genome, and consequently cannot be construed as functional HAV particle receptors but rather binding factors of unknown function. The identification of additional cellular receptors that induce HAV particle conformational changes, as we showed for HAVCR1, will allow a further understanding of the cell entry process of HAV and the role of these receptors in pathogenesis.
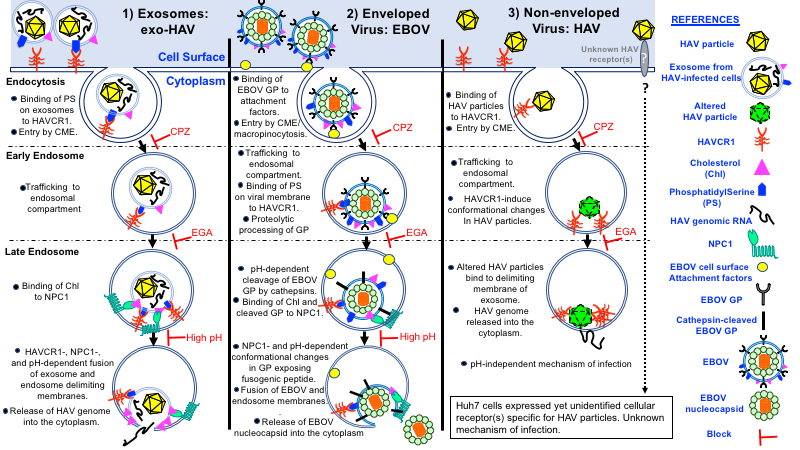
Figure 1. Comparison of cell entry mechanisms by endocytosis of exosomes, enveloped EBOV particles, and non-enveloped HAV particles.Schematic representation of models leading to the release of viral genomes into the cytoplasm. 1) Cargo delivery of exosomes from HAV-infected cells (left panel). Exosomes attach to HAVCR1 at the cell surface, enter the cell by clathrin-mediated endocytosis (CME), traffic from the early endosome (EE) to the late endosome (LE). Cholesterol (Chl) on the exosome membrane binds to NPC1 on the LE membrane resulting in membrane fusion and release of the capsid-free HAV genomic RNA into the cytoplasm. Chlorpromazine (CPZ) that blocks CME, 4-bromobenzaldehyde N-(2,6-dimethylphenyl)semi-carbazone (EGA) that blocks EE to LE transport, and inhibitors of low pH prevented exosome cargo delivery and HAV infection. 2) EBOV infection (middle panel). EBOV enters the cell by CME or macropinocytosis, binds to attachment factors at the cell surface, reaches the EE where HAVCR1 binds to phosphatidylserine(PS) on the viral membrane. Proteases begin to cleave GP, and viral particles traffic from the EE to the LE where cathepsin cleavage GP exposing the NPC1 binding region. Binding of NPC1 to the cleaved GP under low pH triggers conformational changes in GP resulting in the extrusion of the fusogenic peptide, fusion of the delimiting membranes of the virus and LE, and release of the nucleocapsid into the cytoplasm. 3) HAV particle infection (right panel). HAV particles bind to HAVCR1 at the cell surface, enter the cell by CME and are transported to the EE where they bind to more HAVCR1 and undergo conformational changes. The altered HAV capsids bind to the delimiting membrane of the LE, and the HAV genomic RNA is released into the cytoplasm. In contrast to 1 and 2, this process does not require membrane fusion and low pH. Because knockout of HAVCR1 in Huh7 cells blocks cell entry of exosomes from HAV infected cells but not HAV particles, we concluded that these cells express a yet unidentified cellular receptor specific for HAV particles. The cell entry mechanism of HAV particles using this unidentified cellular receptor is unknown and will require further investigation.
By Gerardo Kaplan, Abdolrahim Abbasi, Hsinyi Lu, and Maria Isabel Costafreda
Read the full story at https://www.nature.com/articles/s41564-020-0740-y
References
1 Feng, Z.et al.A pathogenic picornavirus acquires an envelope by hijacking cellular membranes. Nature 496, 367-371, doi:10.1038/nature12029 (2013).
2 Gould, S. J., Booth, A. M. & Hildreth, J. E. The Trojan exosome hypothesis. Proc Natl Acad Sci U S A 100, 10592-10597, doi:10.1073/pnas.1831413100 (2003).
3 Chahar, H. S., Bao, X. & Casola, A. Exosomes and Their Role in the Life Cycle and Pathogenesis of RNA Viruses. Viruses7, 3204-3225, doi:10.3390/v7062770 (2015).
4 Rivera-Serrano, E. E., González-López, O., Das, A. & Lemon, S. M. Cellular entry and uncoating of naked and quasi-enveloped human hepatoviruses. eLife 8, e43983, doi:10.7554/eLife.43983 (2019).
5 Das, A.et al.Gangliosides are essential endosomal receptors for quasi-enveloped and naked hepatitis A virus. Nat Microbiol, doi:10.1038/s41564-020-0727-8 (2020).
6 Kaplan, G.et al.Identification of a surface glycoprotein on African green monkey kidney cells as a receptor for hepatitis A virus. EMBO J 15, 4282-4296 (1996).
7 Feigelstock, D., Thompson, P., Mattoo, P. & Kaplan, G. G. Polymorphisms of the hepatitis A virus cellular receptor 1 in African green monkey kidney cells result in antigenic variants that do not react with protective monoclonal antibody 190/4.J Virol 72, 6218-6222 (1998).
8 Feigelstock, D., Thompson, P., Mattoo, P., Zhang, Y. & Kaplan, G. G. The human homolog of HAVcr-1 codes for a hepatitis A virus cellular receptor. J Virol 72, 6621-6628 (1998).
9 Costafreda, M. I. & Kaplan, G. HAVCR1 (CD365) and its mouse ortholog are functional hepatitis A virus (HAV) cellular receptors that mediate HAV infection. J Virol 92, e02065-02017, doi:10.1128/JVI.02065-17 (2018).
10 Costafreda, M. I. & Kaplan, G. Reply to Das et al., "TIM1 (HAVCR1): an Essential 'Receptor' or an 'Accessory Attachment Factor' for Hepatitis A Virus?". J Virol 93, e02040-02018, doi:10.1128/JVI.02040-18 (2019).
11 Silberstein, E., Dveksler, G. & Kaplan, G. G. Neutralization of hepatitis A virus (HAV) by an immunoadhesin containing the cysteine-rich region of HAV cellular receptor-1. J Virol 75, 717-725, doi:10.1128/jvi.75.2.717-725.2001 (2001).
12 Silberstein, E.et al.Alteration of hepatitis A virus (HAV) particles by a soluble form of HAV cellular receptor 1 containing the immunoglobin-and mucin-like regions. J Virol77, 8765-8774 (2003).
13 Silberstein, E., Konduru, K. & Kaplan, G. G. The interaction of hepatitis A virus (HAV) with soluble forms of its cellular receptor 1 (HAVCR1) share the physiological requirements of infectivity in cell culture. Virol J 6, 175, doi:10.1186/1743-422x-6-175 (2009).
14 Kobayashi, N.et al.TIM-1 and TIM-4 glycoproteins bind phosphatidylserine and mediate uptake of apoptotic cells. Immunity 27, 927-940, doi:10.1016/j.immuni.2007.11.011 (2007).
15 Santiago, C.et al.Structures of T cell immunoglobulin mucin protein 4 show a metal-Ion-dependent ligand binding site where phosphatidylserine binds. Immunity 27, 941-951, doi:10.1016/j.immuni.2007.11.008 (2007).
16 Miyanishi, M.et al.Identification of Tim4 as a phosphatidylserine receptor. Nature 450, 435-439, doi:10.1038/nature06307 (2007).
Follow the Topic
-
Nature Microbiology
An online-only monthly journal interested in all aspects of microorganisms, be it their evolution, physiology and cell biology; their interactions with each other, with a host or with an environment; or their societal significance.





Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in