Virus-inclusive profiling of the human gut microbiome
Published in Microbiology
Explore the Research

Phage-inclusive profiling of human gut microbiomes with Phanta - Nature Biotechnology
Bacteria and viruses are simultaneously quantified in gut metagenomic samples.
In 2021, we were both new members of Ami Bhatt’s translational microbiome laboratory at Stanford. As such, we were both well aware of the vital role that gut microbes play in our health, and eager to make discoveries in this domain. However, we had different primary interests, spanning from the elusive health impacts of gut viruses (Yishay) to the role of the microbiome in neurodegenerative diseases (Meena).
Serendipitously, these interests converged, when in a casual meeting we discovered a mutual interest in studying the changes in the gut virome as people age, and how this compares to changes in the bacteriome. Since most gut viruses are phages, we fondly termed this the “phAging” project and got to work.
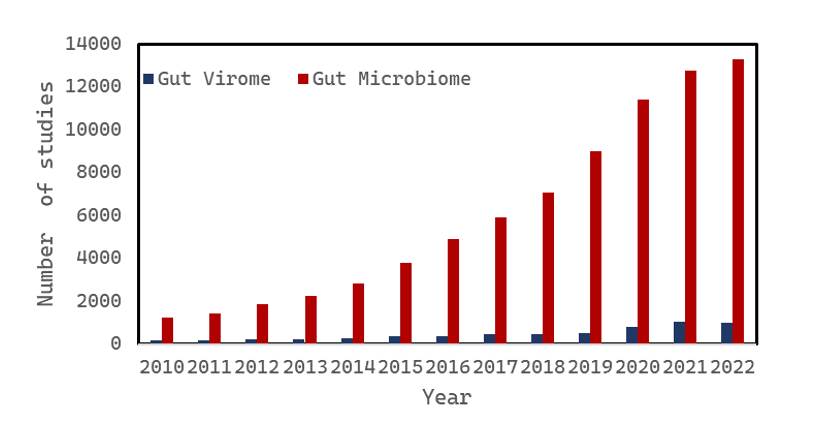
However, shortly after starting our project, we faced a challenge: we realized that existing computational tools to profile metagenomes were not well-suited to simultaneously quantify viruses and bacteria. The reasons for this were primarily technical: for example, many popular tools relied on the existence of reference genomes, which were, until recently, more readily available for bacteria. Tools to identify viruses in metagenomes existed, but were primarily “reference-free,” relying on de novo genome assembly, followed by identification of “virus-like” genomes[1-3]. While assembly-based approaches were crucial in the absence of reference genomes, they are also resource-intensive and have lower sensitivity than reference-based approaches. Thus, many microbiome researchers skipped virome-specific analysis, choosing to stick with the reference-based methods that were optimized for bacterial quantification. Altogether, while the bacterial portion of the human microbiome has been well-studied for over a decade, profiling the viral portion of the microbiome is lagging behind (Figure 1).
We decided to harness the recent surge in availability of reference genomes of viruses from the human gut[4-10], and develop a reference-based, virus-inclusive profiler of human gut metagenomes. This new methodology, now termed “Phanta,” is what we introduce in our recent paper[11]. Phanta (Figure 2) enables fast, tandem profiling of gut viruses and bacteria, directly from short-read shotgun metagenomes. Phanta is freely available on Github (https://github.com/bhattlab/phanta) and can be used to analyze not only new metagenomic datasets, but also the wealth of public datasets that are already available. We hope that its application will reveal novel insights about health conditions that till date have only been studied in relation to gut bacteria.
.png)
Phanta’s first step is classification of short reads from the metagenome to a comprehensive database of human gut microbial genomes. Indeed, the construction of this database was one of the key aspects of developing Phanta, because it includes not only a large collection of prokaryotic genomes present in the human gut, but also a large collection of gut viral genomes, released just a few months before we started our project[4]. Phanta’s database also includes fungal genomes, the human genome, and common “contaminant sequences” in sequencing projects.
With the plethora of viral and prokaryotic reference genomes in the human gut, we decided to adopt Kraken2[12], a k-mer-based approach, for the first step of read classification. However, while benchmarking Phanta, we noticed that even a small fraction of misclassified reads might lead to a relatively high proportion of falsely identified species[13]. To reduce false positives, we introduced a genome coverage-based filtering step (step two). Of note, the default coverage threshold for viral genomes is higher than that for prokaryotes, because viral genomes can be orders of magnitude smaller than prokaryotic ones. Indeed, we take this difference in genome length into account in step 3, before creating the final merged table of viral and prokaryotic abundances.
Using Phanta, we were able to calculate a ratio of ~2:1 DNA virus to bacterial genomes in the healthy adult human gut (n=245 metagenomes from Yachida et al., 2019[14]). We also identified that the most prevalent phages in the gut of these healthy individuals infect Bacteroides, and are either uncharacterized, or recently discovered phages such as Hankyphage[15] and LoVEphage[16].
Phanta offers a valuable platform for profiling phages within microbial communities, particularly in the human gut. Phages play a significant role in human health by directly influencing the gut mucosa and indirectly modulating bacterial population abundance and diversity within the community. Apart from altering bacterial populations, phages can introduce novel functions, such as transferring antibiotic resistance or virulence genes to their bacterial hosts, and can also impact the way the human immune system recognizes the bacterial host. With Phanta, researchers can effectively utilize shotgun metagenomic samples to investigate the interactions between gut viruses and their hosts within the human gut environment, thereby shedding light on their profound effects on human health.
References
- Kieft, K., Zhou, Z. & Anantharaman, K. VIBRANT: automated recovery, annotation and curation of microbial viruses, and evaluation of viral community function from genomic sequences. Microbiome 8, 1–23 (2020).
- Guo, J. et al. VirSorter2: a multi-classifier, expert-guided approach to detect diverse DNA and RNA viruses. Microbiome 9, 1–13 (2021).
- Ren, J. et al. Identifying viruses from metagenomic data using deep learning. Quantitative Biology 8, 64–77 (2020).
- Nayfach, S. et al. Metagenomic compendium of 189,680 DNA viruses from the human gut microbiome. Nat Microbiol 6, 960–970 (2021).
- Gregory, A. C. et al. The Gut Virome Database Reveals Age-Dependent Patterns of Virome Diversity in the Human Gut. Cell Host Microbe 28, 724–740.e8 (2020).
- Soto-Perez, P. et al. CRISPR-Cas System of a Prevalent Human Gut Bacterium Reveals Hyper-targeting against Phages in a Human Virome Catalog. Cell Host Microbe 26, 325–335.e5 (2019).
- Paez-Espino, D. et al. IMG/VR v.2.0: an integrated data management and analysis system for cultivated and environmental viral genomes. Nucleic Acids Res. 47, D678–D686 (2019).
- Tisza, M. J. & Buck, C. B. A catalog of tens of thousands of viruses from human metagenomes reveals hidden associations with chronic diseases. Proc. Natl. Acad. Sci. U. S. A. 118, (2021).
- Camarillo-Guerrero, L. F., Almeida, A., Rangel-Pineros, G., Finn, R. D. & Lawley, T. D. Massive expansion of human gut bacteriophage diversity. Cell 184, 1098–1109.e9 (2021).
- Benler, S. et al. Thousands of previously unknown phages discovered in whole-community human gut metagenomes. Microbiome 9, 78 (2021).
- Pinto, Y., Chakraborty, M., Jain, N. & Bhatt, A. S. Phage-inclusive profiling of human gut microbiomes with Phanta. Nat. Biotechnol. (2023) doi:10.1038/s41587-023-01799-4.
- Wood, D. E., Lu, J. & Langmead, B. Improved metagenomic analysis with Kraken 2. Genome Biol. 20, 257 (2019).
- Breitwieser, F. P., Baker, D. N. & Salzberg, S. L. KrakenUniq: confident and fast metagenomics classification using unique k-mer counts. Genome Biol. 19, 1–10 (2018).
- Yachida, S. et al. Metagenomic and metabolomic analyses reveal distinct stage-specific phenotypes of the gut microbiota in colorectal cancer. Nat. Med. 25, 968–976 (2019).
- Benler, S. et al. A diversity-generating retroelement encoded by a globally ubiquitous Bacteroides phage. Microbiome 6, 191 (2018).
- Van Espen, L. et al. A Previously Undescribed Highly Prevalent Phage Identified in a Danish Enteric Virome Catalog. mSystems 6, e0038221 (2021).
Follow the Topic
-
Nature Biotechnology
A monthly journal covering the science and business of biotechnology, with new concepts in technology/methodology of relevance to the biological, biomedical, agricultural and environmental sciences.



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in