A dual-reference modality to effectively enhance the accuracy of genotyping structural variants from short-read sequencing
Published in Protocols & Methods, Genetics & Genomics, and Mathematics
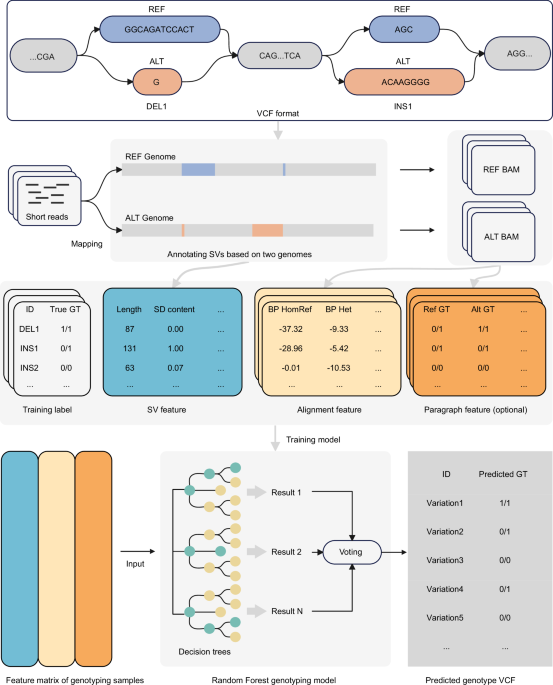
Overview
Structural variants (SVs) are large genomic alterations that play a crucial role in shaping biological traits and contributing to human diseases. Despite advancements in sequencing technologies, accurately genotyping SVs, particularly in repetitive genomic regions, remains a major challenge. In our recent study published in Nature Communications, we introduce SVLearn, a dual-reference-based genotyper designed to enhance SV detection from short-read sequencing data, addressing key limitations in accuracy and cross-species applicability.
Motivation for developing SVLearn
Structural variation is a fundamental driver of genomic diversity, influencing phenotypic traits and disease susceptibility. However, existing SV genotyping methods often struggle with accuracy, particularly in repetitive genomic regions. Many tools either sacrifice computational efficiency or lack the ability to generalize across different species. Our objective was to develop a computational approach that integrates a broad set of genomic, alignment, and genotyping features and leverages a dual-reference strategy to address the above problem. The central idea of the dual-reference strategy is to garner as more information about different kinds of SVs from genomes as possible, significantly increasing the ratio of reads to be mapped to reference genomes. We show that this can improve SV genotyping outcomes in the paper.
Key Findings and Impact
We designed SVLearn to incorporate short-read mapping to both reference and alternative genomes, extracting informative features to train a machine-learning model. By leveraging 38,613 human-derived SVs, we demonstrated that SVLearn achieves up to 15.61% higher precision for insertions and 13.75% higher precision for deletions compared to leading state-of-the-art methods. To assess its generalizability, we validated SVLearn’s performance in cattle and sheep SVs on a large scale, confirming its robust cross-species applicability.
Our approach proved highly effective even at low sequencing coverage. Remarkably, SVLearn maintained genotyping accuracy comparable to 30× coverage using only 5× sequencing depth, making it an invaluable tool for large-scale studies where deep sequencing is not always feasible. This has the potential to accelerate research in genome-wide association studies (GWAS), population genetics, and clinical genomics, providing a more reliable framework for SV genotyping across diverse datasets.
Enroute scheme
While SVLearn represents a significant step forward, there are still challenges to address. The current version focuses on bi-allelic SVs, and future iterations may expand to accommodate more complex variant types, including duplications and inversions. Additionally, integrating SVLearn with more long-read sequencing datasets and graph-based genome representations could further enhance its accuracy and applicability.
Concluding remarks
By developing SVLearn, we hope to empower researchers with a more precise and scalable tool for SV genotyping, paving the way for deeper insights into genomic variation and its implications for health and disease. We are excited to see how the scientific community adopts SVLearn in their research and look forward to collaborating on further advancements in SV analysis, especially how SVs function in ruminants from the evolutionary perspective.
Follow the Topic
-
Nature Communications
An open access, multidisciplinary journal dedicated to publishing high-quality research in all areas of the biological, health, physical, chemical and Earth sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Women's Health
Publishing Model: Hybrid
Deadline: Ongoing
Tumor Microenvironment Crosstalk and Therapeutic Implications
Publishing Model: Hybrid
Deadline: Nov 02, 2026





Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in