A sense-antisense RNA interaction promotes breast cancer metastasis via regulation of NQO1 expression
Published in Cancer

Background
Cancer cells often co-opt post-transcriptional regulatory mechanisms to achieve pathologic expression of cellular pathways that impact metastasis1–3. Previous studies in this field have focused on RNA-binding proteins and micro-RNAs, but antisense RNAs also have significant regulatory potential, as they are ubiquitous in human cells and can base-pair with their sense counterparts4. While, some antisense RNAs have been associated with tumorigenesis5, the extent to which this class of molecules contributes to the regulation of gene expression in cancer remains poorly understood.
Findings
Annotation of antisense RNAs occluded by sense transcripts
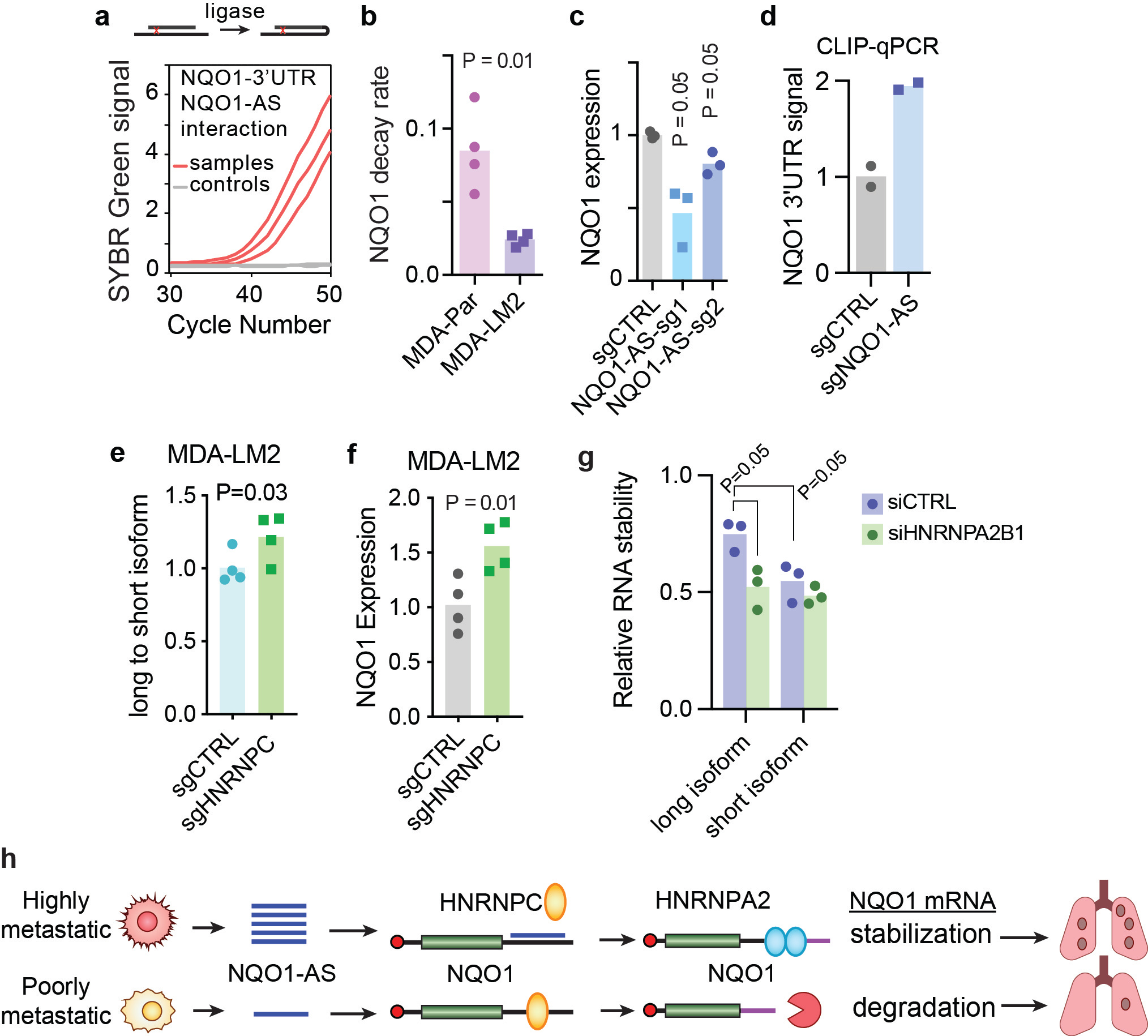
We developed a computational pipeline that integrates data from RNA-seq, global run-on sequencing, and RNA polymerase II ChIP-seq to identify and quantify antisense RNAs. Applying this pipeline to a well-characterized model of breast cancer progression, the MDA-MB-231 parental cell line (MDA-Par) and its highly lung metastatic derivative (MDA-LM2)1,6,7, we identified antisense RNAs significantly upregulated in the highly metastatic line. We focused on one uncharacterized RNA, which we have called NQO1-AS, that is transcribed from the antisense strand of the 3’ UTR of the gene NQO1 (NADPH quinone dehydrogenase 1), and whose sense mRNA is also upregulated in MDA-LM2 cells.
NQO1-AS binds and stabilizes the NQO1 sense transcript
We detected a direct interaction between NQO1-AS and the NQO1 3’UTR using a proximity ligation assay (Fig. 1a). Whole-genome RNA stability measurements1 revealed that NQO1 mRNA is significantly stabilized in MDA-LM2 cells (which upregulate NQO1-AS) relative to the poorly metastatic parental line (Fig. 1b). These findings led us to hypothesize that NQO1-AS binding stabilizes NQO1 post-transcriptionally, and indeed knockdown of NQO1-AS in MDA-LM2 cells caused a decrease in mature NQO1 mRNA (Fig. 1c) with no change in pre-mRNA levels.
NQO1-AS binding modulates polyA site selection and stabilizes the long NQO1 isoform
We hypothesized that NQO1-AS might mask the binding site of a destabilizing factor in the 3’ UTR of NQO1. Analysis of this region revealed an enrichment of motifs recognized by heterogenous nuclear ribonuclear protein C (HNRNPC), and follow-up CLIP-qPCR experiments demonstrated higher HNRNPC binding upon knockdown of NQO1-AS (Fig. 1d).
HNRNPC is a known modulator of polyA site selection8 and NQO1 contains two canonical polyadenylation sites, raising the possibility that HNRNPC binding favors the less stable NQO1 isoform. To test this, we knocked down HNRNPC in MDA-Par cells and observed an increase in the ratio of long to short NQO1 isoforms (Fig. 1e) and in overall NQO1 expression (Fig. 1f), suggesting HNRNPC destabilizes NQO1 by promoting the formation of the truncated isoform.
To determine the mechanism by which the long and short NQO1 isoforms are differentially regulated, we searched CLIP-seq datasets for motifs present only in the longer isoform, and we found that the extended 3’UTR contains several binding sites for the RNA binding protein HNRNPA2B1, which has been shown to act as a stabilizing factor9. Separate measurement of the long and short NQO1 isoform decay rates revealed that the long isoform is indeed significantly more stable, and that this difference in stability is abrogated when HNRNPA2B1 is silenced (Fig. 1g).
Altogether, our results suggest that NQO1-AS overexpression in highly metastatic breast cancer cells decouples NQO1 from the broader HNRNPC regulon, enabling NQO1 upregulation via a stabilizing interaction with HNRNPA2B1 (Fig. 1h).
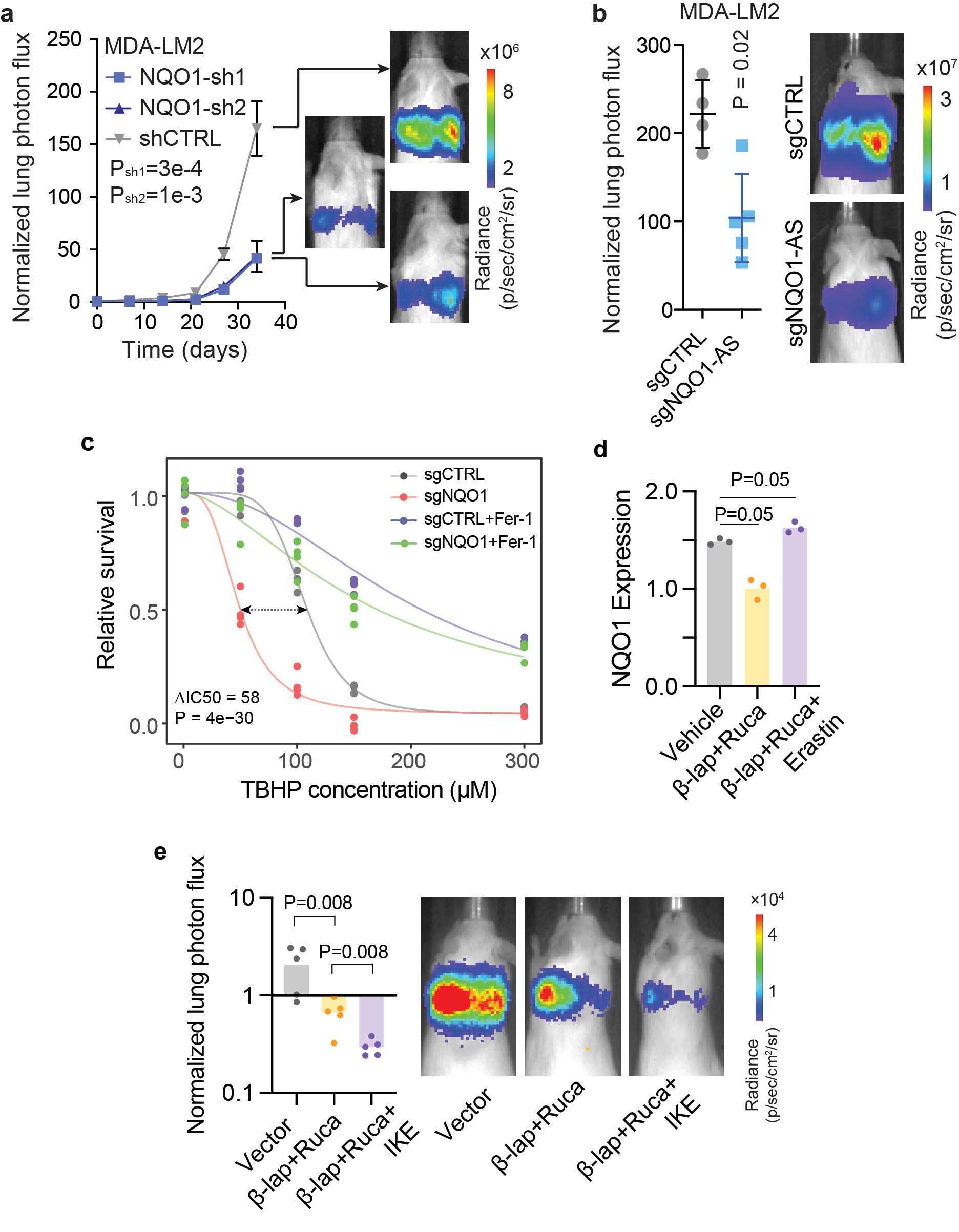
NQO1 and NQO1-AS promote metastatic lung colonization
We next investigated the impact of NQO1 upregulation on breast cancer progression by performing in vivo lung colonization assays with NQO1/NQO1-AS knockdown and control MDA-LM2 cells. While knocking down NQO1 or NQO1-AS did not affect the in vitro proliferation rate of the cells, it significantly decreased their lung colonization (Fig. 2a,b), indicating that the overexpression of these molecules enhances the capacity of breast cancer cells to metastasize to the lung.
NQO1 protects cancer cells from ferroptosis
NQO1 is a chemoprotective enzyme involved in cellular defense against oxidizing agents10, and ferroptosis has been shown to be a consequence of excessive oxidative stress in metastatic breast cancer11,12. We therefore hypothesized that NQO1 upregulation may be a mechanism for breast cancer cells to protect themselves from ferroptosis. Consistent with this hypothesis, NQO1 knockdown cells were significantly more sensitive than controls to treatment with various ferroptosis inducers, including tert-butyl hydroperoxide (Fig. 2c). Treatment with Ferrostatin-1, an inhibitor of ferroptosis13, rescued ferroptosis inducer-mediated toxicity. Importantly, NQO1 knockdown did not sensitize cells to apoptosis, necroptosis, or autophagy, indicating that its protective role is at least somewhat specific to ferroptosis.
The NQO1/NQO1-AS pathway can be exploited therapeutically
The compound β-Lapachone is metabolized by NQO1 into an unstable hydroquinone that spontaneously generates superoxide14, leading to programmed necrosis of cancer cells15. β-Lapachone has proved effective in cancers with increased NQO1 expression; however, its sustained use at high concentrations causes anemia in both humans and animal models16. Combining β-Lapachone with poly(ADP-ribose) polymerase (PARP) inhibitors results in synergistic anti-tumor activity17, and we hypothesized that adding a ferroptosis inducer could further increase the potency of this therapeutic regimen. This hypothesis was in part based on the observation that MDA-Par cells exhibit a high degree of heterogeneity in NQO1 expression, so targeting PARP and NQO1 while inducing ferroptosis could simultaneously kill subpopulations of cancer cells with high and low NQO1 expression. To test this, we treated MDA-LM2 cells with either β-Lapachone + Rucaparib (a PARP inhibitor), Erastin, or vehicle control and measured NQO1 expression in the surviving cells (Fig. 2d). We found that the cells that survived β-Lapachone + Rucaparib treatment had relatively low NQO1, whereas the cells that survived Erastin treatment had relatively high NQO1, indicating that these treatments target distinct subpopulations. To test our proposed combination therapy in vivo, we conducted a lung colonization assay with MDA-Par cells and found that the addition of IKE improved the efficacy of β-Lapachone + Rucaparib treatment (Fig. 2e). This suggests that the role of NQO1 as a protective agent against ferroptosis can be exploited by treatment with a ferroptosis inducer in combination with established NQO1-inhibitor therapies.
Conclusion
Here, we developed a pipeline to profile antisense RNAs and applied it to an established model of breast cancer metastasis. We identified NQO1-AS, whose upregulation promotes breast cancer metastasis. Taken together, our data supports a model in which NQO1-AS binds to the 3’ UTR of NQO1, preventing the binding of HNRNPC, and thereby favoring the use of the distal polyadenylation site. HNRNPA2B1 can then bind to the long 3’UTR, stabilizing the mRNA and increasing the level of the NQO1 enzyme. Breast cancer cells exploit this pathway during metastatic progression, enabling them to tolerate higher levels of oxidative stress, and cells that successfully colonize the lungs become dependent on this pathway for survival, making them vulnerable to therapies that target this pathway. In preliminary experiments, we have shown that combining NQO1 and PARP inhibitors with a ferroptosis inducing agent can significantly reduce metastatic burden in vivo.
References
- Goodarzi, H. et al. Metastasis-suppressor transcript destabilization through TARBP2 binding of mRNA hairpins. Nature 513, 256–260 (2014).
- Goodarzi, H. et al. Endogenous tRNA-Derived Fragments Suppress Breast Cancer Progression via YBX1 Displacement. Cell 161, 790–802 (2015).
- Goodarzi, H. et al. Modulated Expression of Specific tRNAs Drives Gene Expression and Cancer Progression Article Modulated Expression of Specific tRNAs Drives Gene Expression and Cancer Progression. Cell 165, 1416–1427 (2016).
- Ozsolak, F. et al. Comprehensive Polyadenylation Site Maps in Yeast and Human Reveal Pervasive Alternative Polyadenylation. Cell 143, 1018–1029 (2010).
- Balbin, O. A. et al. The landscape of antisense gene expression in human cancers. Genome Res. 25, 1068–1079 (2015).
- Minn, A. J. et al. Genes that mediate breast cancer metastasis to lung. Nature 436, 518–524 (2005).
- Tavazoie, S. F. et al. Endogenous human microRNAs that suppress breast cancer metastasis. Nature 451, 147–152 (2008).
- Navickas, A. et al. An mRNA processing pathway suppresses metastasis by governing translational control from the nucleus. Nature Cell Bio in press (2021).
- Goodarzi, H. et al. Systematic discovery of structural elements governing stability of mammalian messenger RNAs. Nature 485, 0–6 (2012).
- Ross, D. Quinone Reductases Multitasking in the Metabolic World. Drug Metab. Rev. 36, (2004).
- Dixon, S. J. & Stockwell, B. R. The Hallmarks of Ferroptosis. Annu. Rev. Cancer Biol. 3, 35–54 (2019).
- Bi, J. et al. Metadherin enhances vulnerability of cancer cells to ferroptosis. Cell Death Dis. 10, (2019).
- Zilka, O. et al. On the Mechanism of Cytoprotection by Ferrostatin-1 and Liproxstatin-1 and the Role of Lipid Peroxidation in Ferroptotic Cell Death. ACS Cent. Sci. 3, 232–243 (2017).
- Reinicke, K. E. et al. Development of beta-lapachone prodrugs for therapy against human cancer cells with elevated NAD(P)H:quinone oxidoreductase 1 levels. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 11, 3055–3064 (2005).
- Huang, X. et al. An NQO1 substrate with potent antitumor activity that selectively kills by PARP1-induced programmed necrosis. Cancer Res. 72, 3038–3047 (2012).
- Noh, J.-Y. et al. A naphthoquinone derivative can induce anemia through phosphatidylserine exposure-mediated erythrophagocytosis. J. Pharmacol. Exp. Ther. 333, 414–420 (2010).
- Huang, X. et al. Leveraging an NQO1 Bioactivatable Drug for Tumor- Selective Use of Poly ( ADP-ribose ) Polymerase Inhibitors. Cancer Cell 30, 940–952 (2016).
Follow the Topic
-
Nature Cancer
This journal aims to provide a unique forum through which the cancer community will learn about the latest, most significant cancer-related advances across the life, physical, applied and social sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Cancer Neuroscience: from mechanisms to therapy
Publishing Model: Hybrid
Deadline: Jan 30, 2027


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in