AF1q is a universal marker of neuroblastoma that sustains N-Myc expression and drives tumorigenesis
Published in Cancer and Paediatrics, Reproductive Medicine & Geriatrics
What is neuroblastoma, and why is it important?
Neuroblastoma is the most common extracranial solid tumor of childhood, and although it accounts for only 6% of childhood malignancies based on incidence (Figure 1), it is responsible for 15% of childhood cancer deaths 1. It develops from neural crest, an embryonic tissue that gives rise to sensory, sympathetic, and parasympathetic neurons. Some neuroblastoma tumors spontaneously regress or differentiate, and low-risk neuroblastomas are associated with a 90-95% cure rate 2. However, neuroblastoma can also present in an aggressive form that is relentless and resistant to multimodal therapy including high-dose myeloablative chemotherapy, radiation and stem-cell transplantation 3. High-risk neuroblastomas are associated with a less than 50% five-year event-free survival rate, making identification of novel and more effective therapies a critical goal.

What are Myc proteins, and what role do they play in neuroblastoma?
Myc family proteins are fundamental to the biology of neuroblastoma 4. Amplification of MYCN is observed in ~25% of neuroblastomas and is associated with unfavorable outcome. Augmented MYC expression is responsible for aggressive behavior in other neuroblastomas 5. Myc proteins are basic helix-loop-helix and leucine zipper transcription factors that act through transcriptional and target-gene independent mechanisms to control cell proliferation, apoptosis, metabolism and other cellular processes whose regulation undergirds normal homeostasis and whose deregulation is fundamental to carcinogenesis 6.
How are Myc proteins regulated within the cell?
Under physiological conditions, Myc proteins are short-lived with a half-life of 15-30 minutes due to their rapid proteasomal degradation [7]. The AKT/GSK3β and Ras/ERK signaling pathways mediate two separate protein phosphorylation cascades that terminate in the Myc proteins themselves, ultimately controlling their ubiquitination on conserved lysine residues, thereby determining their fate [8]. To date, 18 ubiquitin ligases, 6 deubiquitinating enzymes and one SUMO protease have been shown to control ubiquitination and stability of Myc proteins [9]. In response to physiological growth signals, the half-lives of Myc proteins may be temporarily extended via interactions impinging upon AKT/GSK3β and/or Ras/ERK pathways or ubiquitin posttranslational modifications [8]. However, in neuroblastoma N-Myc or C-Myc protein expression levels are sustained through a variety of mechanisms. This pathway represents a potentially exploitable vulnerability in neuroblastoma.
What is AF1Q, and what is its role in malignancies?
AF1Q (ALL1-fused gene from chromosome 1q) also known as MLLT11, encodes a 90-amino-acid protein (AF1q) with no known enzymatic activity. AF1q was first identified in an infant with acute myelogenous leukemia (AML) harboring a chromosomal translocation and was subsequently found to be overexpressed in hematologic malignancies as well as in several solid tumors 7-14. In most of these malignancies, AF1q expression is a predictor of poor prognosis. In various cancer cell model systems, AF1q upregulation has been associated with enhanced proliferation, migration, and invasion 10,11,15-17. Moreover, AF1q has been shown to activate the AKT pathway as well as being a component of the Wnt pathway by acting as a co-factor for TCF7 11,18. AF1q is expressed primarily in neural crest-cell derived ganglia 19. Ectopic AF1q expression can also promote neuronal differentiation in non-neuronal cells 20. Despite its neurogenic properties and expression pattern, to our knowledge, a role for AF1q in neural tumors has not been reported. In this study, we explored the potential role of AF1q in neuroblastoma.
Results
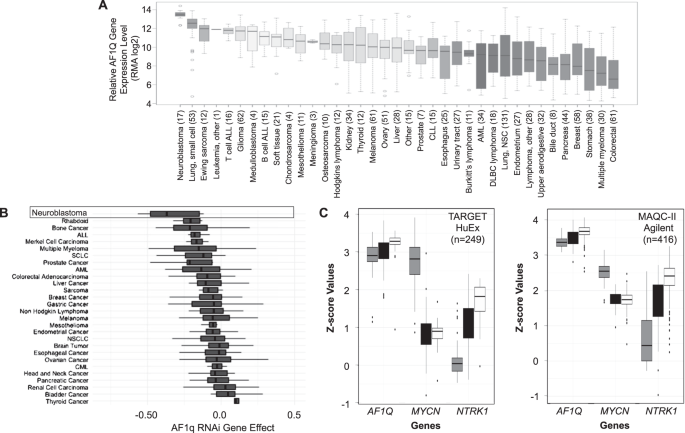
Using the Broad Institute’s Cancer Cell Line Encyclopedia database to compare AF1Q gene expression in 37 different types of pediatric and adult malignancies, we found that AF1Q was expressed at the highest levels in neuroblastoma compared to all other tumor types (Figure 2).

Using the “Depmap” Cancer Dependency Map database to analyze the impact of gene silencing and editing of different cancer cell lines, we found that neuroblastoma cells were also more reliant upon AF1Q than any other cell line. In addition, AF1q was universally expressed in human neuroblastoma tumors regardless of risk classification.
Importantly, silencing AF1q in neuroblastoma cells resulted in cell cycle arrest, apoptosis, and a profound reduction in tumorigenicity based on the ability of neuroblastoma cell xenografts to form tumors in immunodeficient mice (Figure 3).

Figure 3. In vivo tumorigenicity of neuroblastoma cells requires AF1q expression. Tumor bioluminescence of AF1q knockdown (AF1q KD) and control Kelly cell xenografts imaged at 30 days after implantation into nude mice.
Moreover, through a series of cell biology experiments in multiple neuroblastoma cell lines, we demonstrated that AF1q protects N-Myc from ubiquitination and proteasomal degradation. It does so by impacting AKT/GSK3b and Ras/ERK signaling pathways.
Conclusion
Our overall findings reveal AF1q to be a novel regulator of N-Myc and a driver of neuroblastoma viability and tumorigenicity. A proposed model of AF1q’s role in neuroblastoma is shown in Figure 4.

Figure 4. Schematic model of AF1q’s effect in neuroblastoma. Two different patterns of cell signaling activity are present in neuroblastoma cells when AF1q is expressed or silenced. (A) In the presence of AF1q, AKT is active, and its substrate GSK3b is phosphorylated, rendering it inactive. ERK is active and phosphorylates N-Myc on serine 62, stabilizing it. MDM2 levels are high (possibly due to its known upregulation by N-Myc). MDM2 promotes the proteasomal degradation of p53. These conditions prevent apoptosis and promote tumorigenesis. (B) In the absence of AF1q, AKT is inactive, leaving GSK3b unphosphorylated and active and therefore able to phosphorylate N-Myc on threonine 58. Further, in the absence of AF1q, Ras does not activate ERK, leaving N-Myc serine 62 unphosphorylated. These conditions render N-Myc susceptible to proteasomal degradation. MDM2 levels are low (possibly due to loss of N-Myc), and p53 is upregulated. Through an unknown mechanism, p53 is activated in the absence of AF1q and stimulates mitochondrial apoptosis, which is evident by BAX activation.
References
1. Zafar A, Wang W, Liu G, et al. Molecular targeting therapies for neuroblastoma: Progress and challenges. Med Res Rev. 2021;41(2):961-1021.
2. Colon NC, Chung DH. Neuroblastoma. Advances in Pediatrics. 2011;58(1):297-311.
3. Tolbert VP, Matthay KK. Neuroblastoma: clinical and biological approach to risk stratification and treatment. Cell Tissue Res. 2018;372(2):195-209.
4. Beltran H. The N-myc oncogene: maximizing its targets, regulation, and therapeutic potential. Mol Cancer Res. 2014;12(6):815-822.
5. Wang LL, Teshiba R, Ikegaki N, et al. Augmented expression of MYC and/or MYCN protein defines highly aggressive MYC-driven neuroblastoma: a Children's Oncology Group study. Br J Cancer. 2015;113(1):57-63.
6. Baluapuri A, Wolf E, Eilers M. Target gene-independent functions of MYC oncoproteins. Nat Rev Mol Cell Biol. 2020;21(5):255-267.
7. Strunk CJ, Platzbecker U, Thiede C, et al. Elevated AF1q expression is a poor prognostic marker for adult acute myeloid leukemia patients with normal cytogenetics. Am J Hematol. 2009;84(5):308-309.
8. Tse W, Zhu W, Chen HS, Cohen A. A novel gene, AF1q, fused to MLL in t(1;11) (q21;q23), is specifically expressed in leukemic and immature hematopoietic cells. Blood. 1995;85(3):650-656.
9. Tiberio P, Lozneanu L, Angeloni V, et al. Involvement of AF1q/MLLT11 in the progression of ovarian cancer. Oncotarget. 2017;8(14):23246-23264.
10. Chang XZ, Li DQ, Hou YF, et al. Identification of the functional role of AF1Q in the progression of breast cancer. Breast Cancer Res Treat. 2008;111(1):65-78.
11. Hu J, Li G, Liu L, Wang Y, Li X, Gong J. AF1q mediates tumor progression in colorectal cancer by regulating AKT signaling. Int J Mol Sci. 2017;18(5).
12. Gruber ES, Oberhuber G, Birner P, et al. The oncogene AF1Q is associated with WNT and STAT signaling and offers a novel independent prognostic marker in patients with resectable esophageal cancer. Cells. 2019;8(11).
13. Co NN, Tsang WP, Tsang TY, et al. AF1q enhancement of gamma irradiation-induced apoptosis by up-regulation of BAD expression via NF-kappaB in human squamous carcinoma A431 cells. Oncol Rep. 2010;24(2):547-554.
14. Busson-Le Coniat M, Salomon-Nguyen F, Hillion J, Bernard OA, Berger R. MLL-AF1q fusion resulting from t(1;11) in acute leukemia. Leukemia. 1999;13(2):302-306.
15. Park J, Kim S, Joh J, et al. MLLT11/AF1q boosts oncogenic STAT3 activity through Src-PDGFR tyrosine kinase signaling. Oncotarget. 2016;7(28):43960-43973.
16. Jin H, Sun W, Zhang Y, et al. MicroRNA-411 Downregulation Enhances Tumor Growth by Upregulating MLLT11 Expression in Human Bladder Cancer. Mol Ther Nucleic Acids. 2018;11:312-322.
17. Li W, Ji M, Lu F, et al. Novel AF1q/MLLT11 favorably affects imatinib resistance and cell survival in chronic myeloid leukemia. Cell Death Dis. 2018;9(9):855.
18. Park J, Schlederer M, Schreiber M, et al. AF1q is a novel TCF7 co-factor which activates CD44 and promotes breast cancer metastasis. Oncotarget. 2015;6(24):20697-20710.
19. Yamada M, Clark J, Iulianella A. MLLT11/AF1q is differentially expressed in maturing neurons during development. Gene Expr Patterns. 2014;15(2):80-87.
20. Lin HJ, Shaffer KM, Sun Z, Jay G, He WW, Ma W. AF1q, a differentially expressed gene during neuronal differentiation, transforms HEK cells into neuron-like cells. Brain Res Mol Brain Res. 2004;131(1-2):126-130.
Follow the Topic
-
Oncogene
This journal aims to make substantial advances in our knowledge of processes that contribute to cancer by publishing outstanding research.





Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in