After the Paper | From Paper to Industrial-scale Platform: a 3-Year Behind the Paper Journey from GENTRL to Chemistry42
Published in Bioengineering & Biotechnology
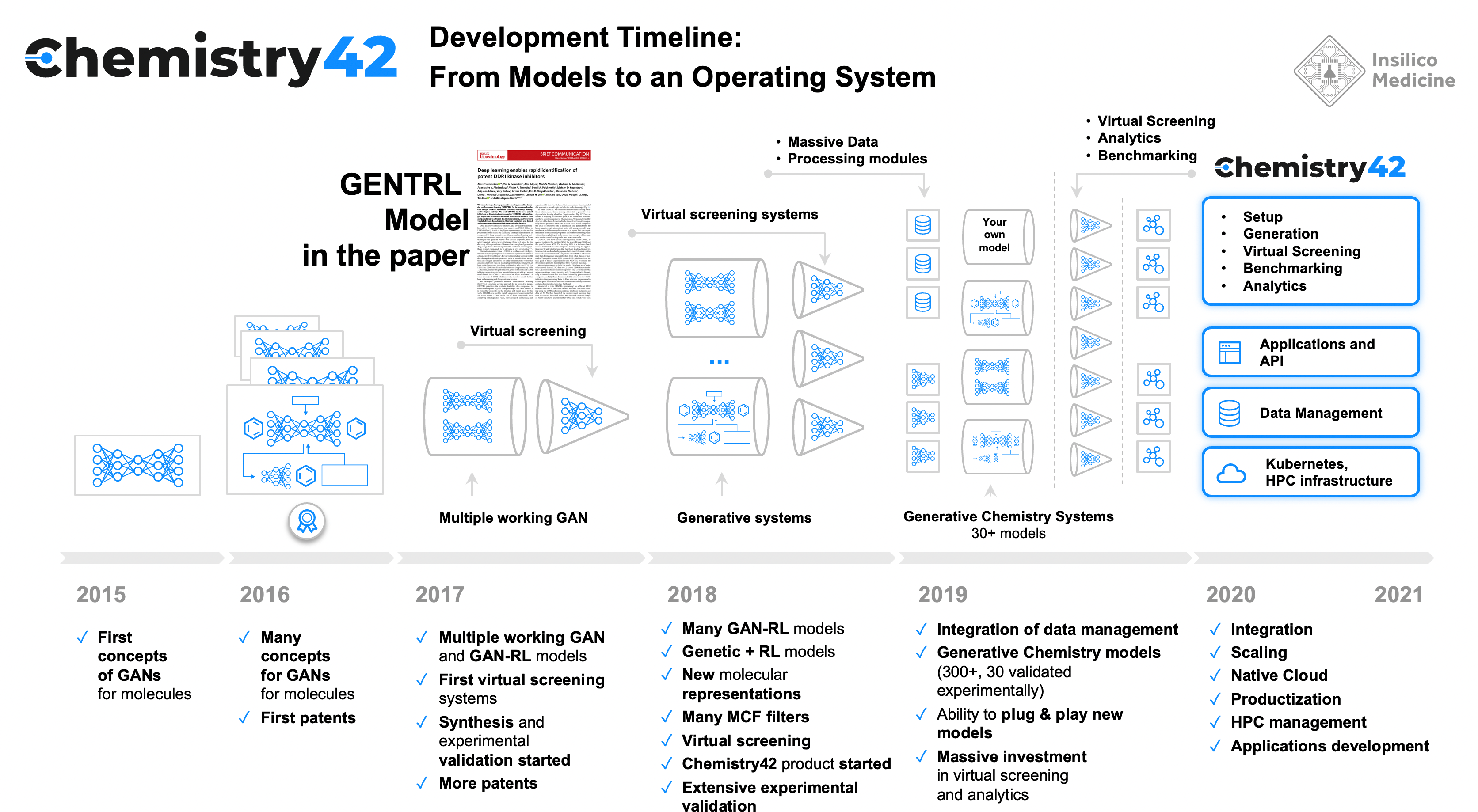

In September 2019, after almost a year in review, our paper titled “Deep learning enables rapid identification of potent DDR1 kinase inhibitors” was published in Nature Biotechnology. This paper was the result of a “demo race” where WuXi AppTec, the world’s largest platform for drug discovery R&D, tested one of the generative chemistry models developed by Insilico Medicine called the Generative Tensorial Reinforcement Learning (GENTRL - developed in 2018). In 2018, the Insilico team was challenged to use this model to come up with molecules that could be synthesized and tested in record time. The paper immediately drew the attention of researchers in the field. Just a week after publication, it was among the top eight most popular papers in the history of Nature Biotechnology by the Altmetric Score, a measure of public attention.
The story behind the paper was published in another Behind the Paper article on Nature Bioengineering. Here is the story of what happened three years after the paper was published. A variation of this model was later presented at NeurIPS in 2019.
Before GENTRL, Insilico scientists published a number of papers on generative chemistry using GANs and other generative approaches. In 2017, we published our work on the design of a selective JAK inhibitor using AI. In 2018 another prominent group led by Gisbert Schneider also published experimental data related to AI-designed molecules.(Ref) Insilico was also competing with the most prominent academic group in the field led by, Alán Aspuru-Guzik’s at Harvard. In 2016, while Insilico’s paper titled “The Cornucopia of Meaningful Leads” was in review at a peer-reviewed journal, Alan and his team published the “Automated Chemical Design” paper on the preprint server (later published in ACS Central Science) and got a lot of attention. e admired Aspuru-Guzik’s group and shared his mission to generate and synthesize molecules using machine learning, ultimately partnering with Alán and becoming long-term collaborators.

The review process of GENTRL was challenging. At that time, there were very few groups working on the intersection of algorithm design and drug discovery. Classical drug hunters did not care about the beauty of the algorithm and algorithm designers did not understand the fundamentals of drug discovery. In addition, many of the groups, AI or not, were competing with each other, and some of the early adopters were going public.
After our paper was published, a group from Relay Therapeutics, a company using the Schrödinger CADD physics-based platforms on a powerful computer and in which Schrödinger had a substantial stake, criticized the paper. Together with Alán, we responded. One of the criticisms was that out of the 4 molecules that were synthesized and tested in in vitro assays, the one that was tested in mice was similar to ponatinib. Nowadays, most users of generative chemistry platforms understand that one of the limiting factors is budget for synthesis and testing. When the budget is low, you prioritize the molecules that are cheaper to synthesize. In addition, it is important to note that while the molecule may have been similar to Ponatinib, it was novel, and therefore, patentable.
But, needless to say, that debate led to a very important realization - we had to turn GENTRL into a tool that others could use to design novel molecules that can be synthesized and turned into effective drugs. 
From a business perspective, we needed to focus on software development.There are a number of platform companies that claim that they use AI to develop their own pipeline of therapeutics without revealing or sharing their AI tools with the market. Software businesses are generally not as attractive to investors because the total available market for software is very small and is dominated by companies with large sales teams and many years on the market. And so far, one algorithm alone or one approach can not take over all of the drug discovery tasks. As we can see from the history of AlphaFold, which was first used in 2018, there are no molecules in the clinic where AlphaFold was used at the start of the program. That being said, Insilico Medicine is working on another partnership with Alán Aspuru-Guzik’s group, where we have set the first such example.

Chemistry42: From Research Papers to the Industrial-strength Multi-AI Platform
In 2019, we endeavored to develop an AI tool that can be used by the most sophisticated teams in the pharmaceutical industry. These companies were also developing their own AI systems. And GENTRL alone would not be sufficient.
By 2019, the pharmaceutical industry had already developed substantial internal capabilities in AI and we knew that in order to have any impact, we needed to develop tools that were very customizable.

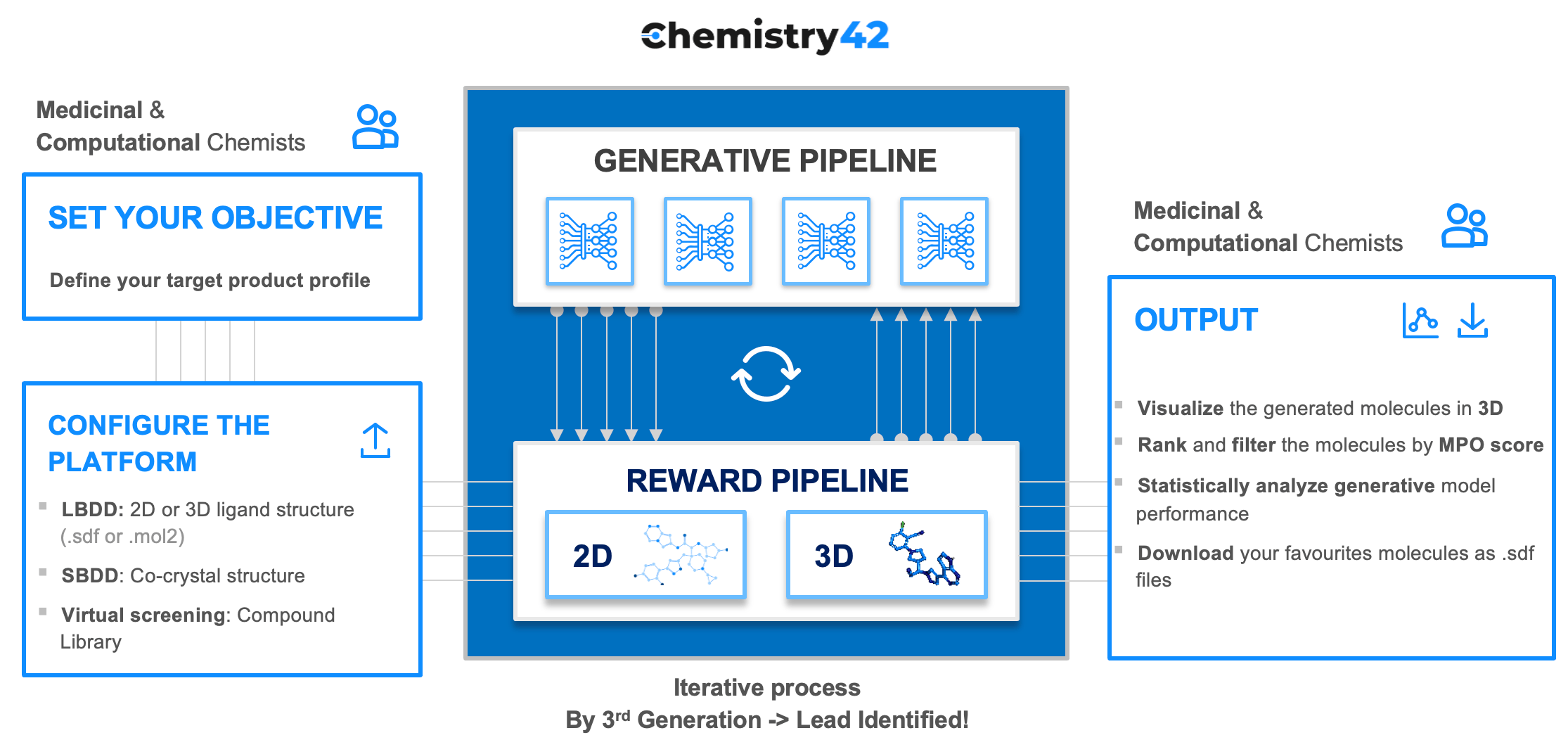
Unfortunately, one generative model alone can not yet produce novel molecules with the desired properties that can be synthesized and reach human clinical trials. Such an effort requires multiple generative models, a large number of predictive models and a reinforcement learning system where the output of each individual model and an ensemble is evaluated and the models and ensemble is rewarded or punished for generating the molecules with desired properties. We designed Chemistry42 to have a user friendly interface and intuitive workflow that requires only 3 steps.
- Step 1. Set your objective
- Step 2. Configure the platform with the criteria you wish the compounds to satisfy, and let the platform run for between 2 - 72 hours
- Step 3. Visualize the generated compounds and rank and filter them based on your preferred criteria
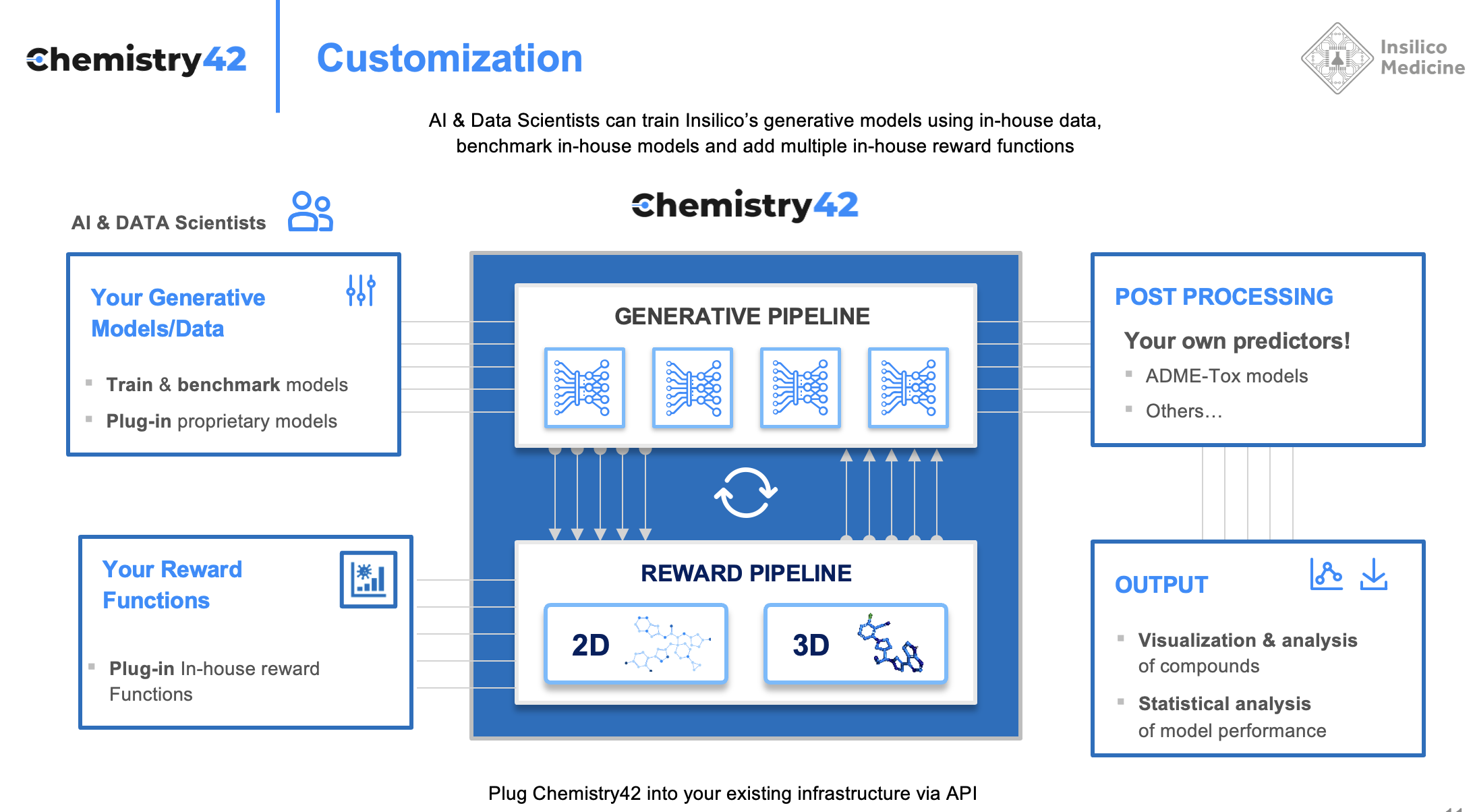
In addition, since the many pharmaceutical companies have their own AI groups that also specialize in generative chemistry and want to get the tools that allow them to rapidly benchmark and train their own models as well as to get the tools that allow to pick the best generated molecules, we had to allow for platform customization. For hit and lead optimization strategies, project specific predictive models can be added to the reward pipeline. Companies with their own in-house generalizable models can also customize the platform to include them in the reward pipeline.
We had to allow the scientists to add their own generative models or use their own training data. This customization and flexibility allowed us to go outside drug discovery and in agro chemistry and even into green chemistry.
Beyond Molecule Prediction – End to End AI for Drugs With Human Patients in Mind
We recognized that GENTRL, like AlphaFold, is only a piece of the puzzle. We sought to create an end-to-end AI platform–Pharma.AI–that could bridge the gap between predicting novel structures and producing actual drugs that help patients. And there are very many pieces of the drug discovery puzzle where AI can help.
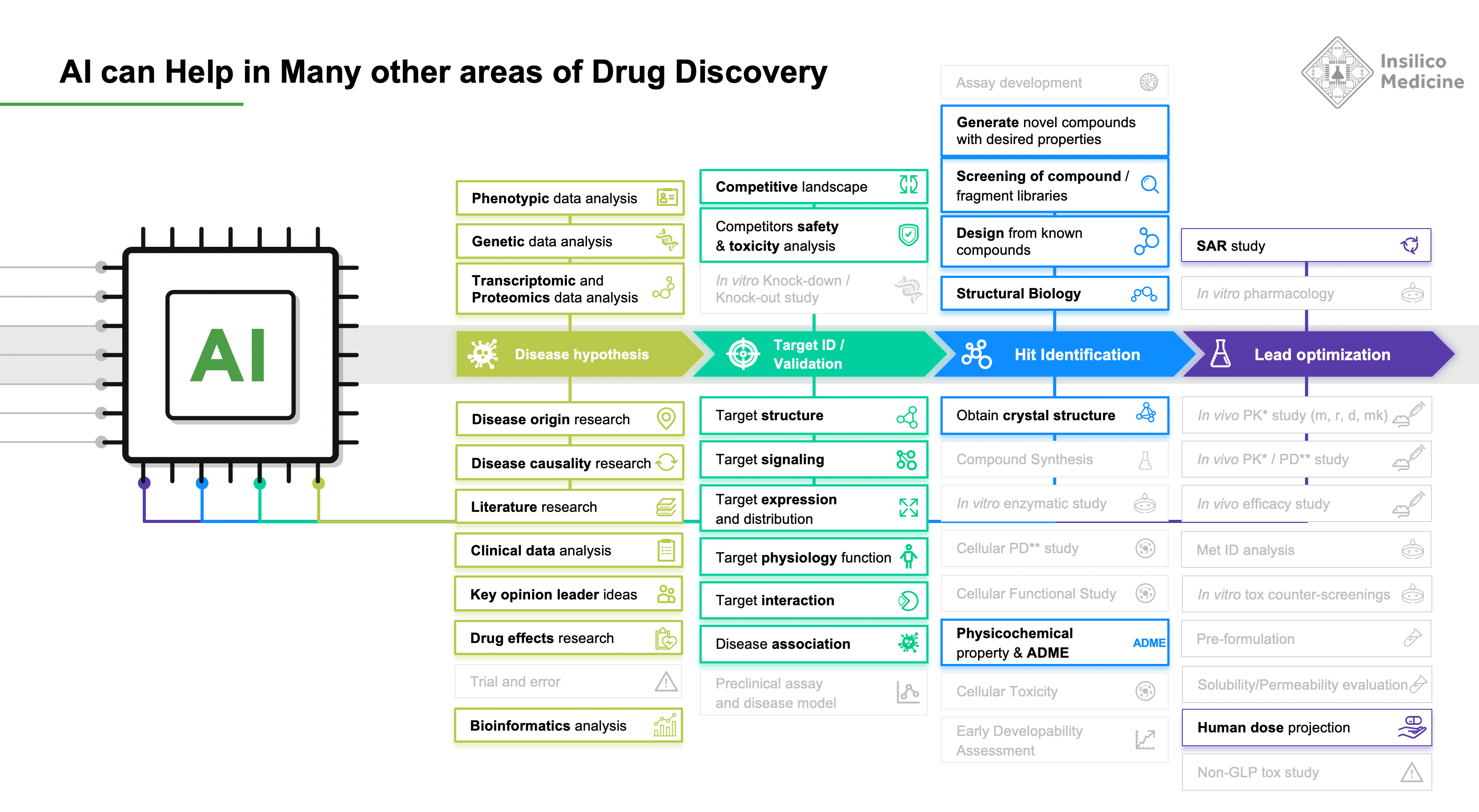
As chemist and author Derek Lowe writes in Chemistry World, predicting protein structures as AlphaFold does not itself revolutionize drug discovery. Part of it has to do with the structural weirdness of certain proteins – those with “disordered protein regions” have no existing structure for a computer to use as a comparison. And part of the problem is that these proteins are still unvalidated predictions. In the end, writes Lowe, small molecules made from protein structures have all sorts of problems that lead to failures in the clinic and rarely do those problems have to do with the structure of the target. He writes: “In the end the real numbers from the real biological system are what matter. As a project goes on, those numbers include assays covering pharmacokinetics, metabolism, and toxicology, and none of those can really be dealt with from the level of protein structure.”
Using AI to Improve Drug Discovery End to End
In order to ensure that these computer-generated molecules work in actual, biologically complex organisms, Insilico Medicine designed an end-to-end AI platform that connects target identification with small molecule design. This platform is validated through testing the most promising lead candidates, and running parallel experiments that feed all data and results back into its AI system to continually improve its output and predictive capabilities. The platform is called Pharma.AI, and recently the company has taken steps to improve the outcome of clinical trials through the development of an additional AI system called InClinico.
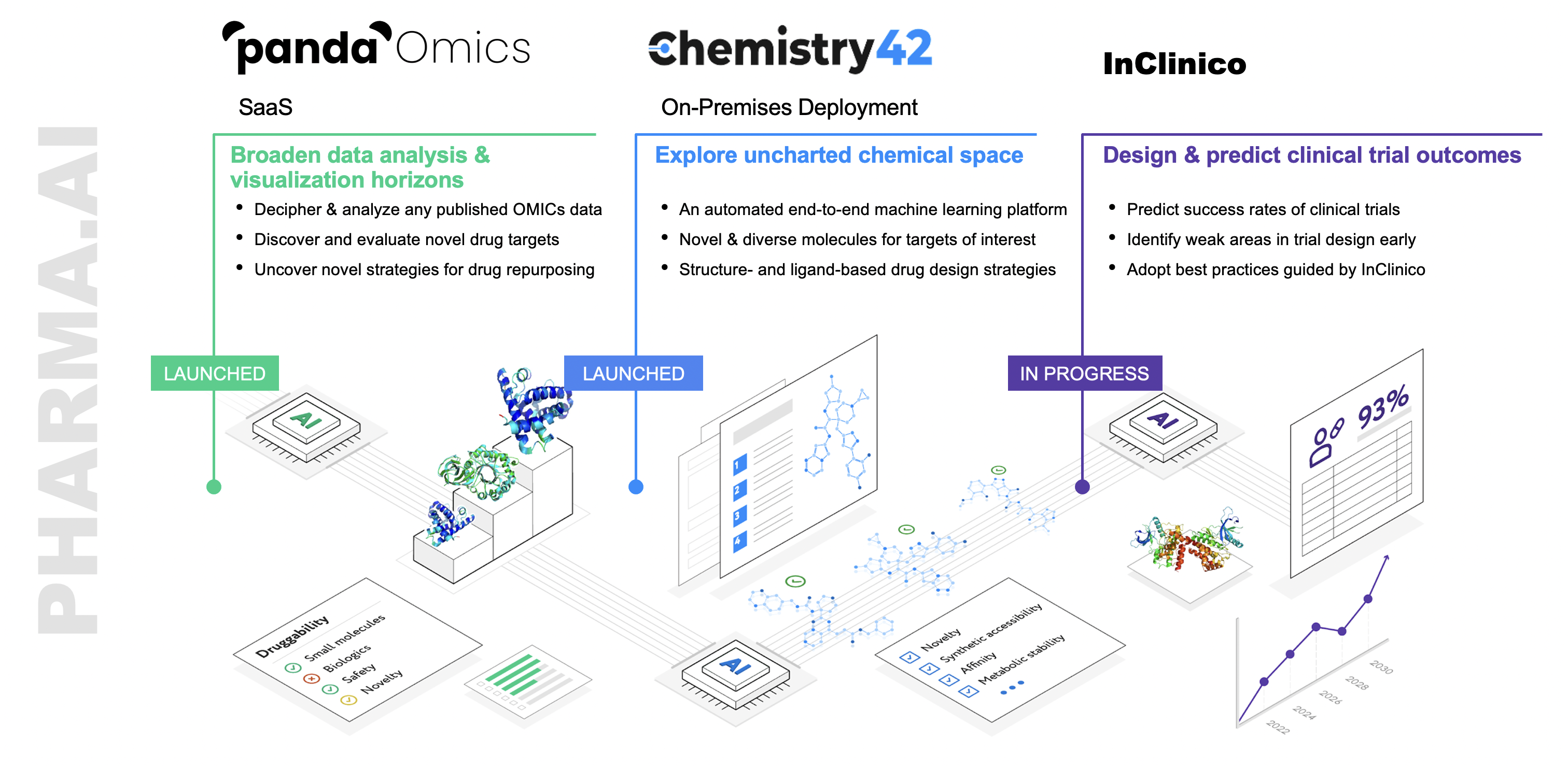
But first, the chemistry. In 2020, building off of our successful work with GENTRL, we released Chemistry42, a small-molecule generating AI platform that can design, rank and score millions of compounds to find hundreds with desired properties—whether those are existing drugs or potentially new therapeutics. Chemistry42’s AI is trained on 10 million publicly available compounds, and 100 million building blocks—or virtual molecular fragments.
With a speed and efficiency that far surpasses the ability of human scientists, and proven results as evidenced by Insilico’s own internal pipeline programs, there was no question that the technology worked, and pharmaceutical companies began turning to the Chemistry42 platform to improve their own searches for the next breakthrough therapies.
To design these promising novel molecules, the generative engine of the Chemistry42 platform generates hundreds of molecular structures that are funneled into a reward pipeline. This reward pipeline assesses each structure’s suitability and selects high-scoring molecules, those that meet objectives such as safety, potency, synthetic availability, and metabolic stability. The generated molecules and their subsequent scores are returned to the generative engine so that the models “learn” the types of molecules that score highly and those that score poorly. Based on these data, the generative models are re-trained to generate high scoring molecules.
Validating End-to-End AI Drug Discovery Capabilities
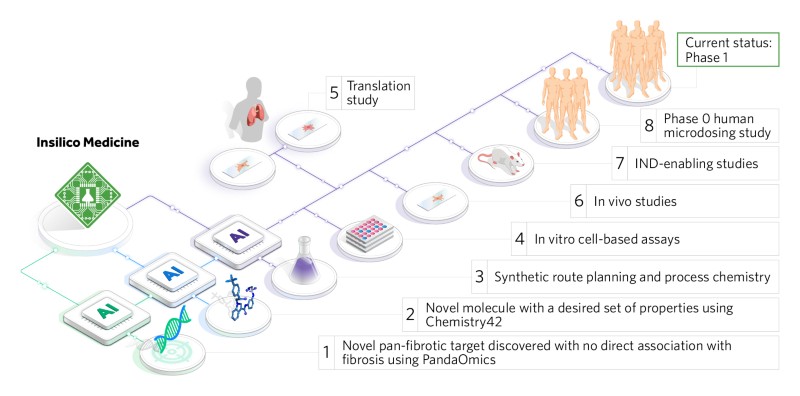
Insilico’s Pharma.AI platform has managed to go far beyond mere prediction–with the release of a number of preclinical candidates that have progressed to later stage studies, most notably a lead candidate for treating idiopathic pulmonary fibrosis. In under 30 months, and at a fraction of the cost of traditional drug development, we brought a completely AI-discovered and AI-designed novel drug to Phase 1 trials.
https://www.nature.com/articles/d43747-021-00039-5
Here’s how we did it. The target discovery system, PandaOmics, identified targets through deep feature selection, causality inference and de novo pathway reconstruction. We then used a natural language processing (NLP) engine to assess the targets’ novelty and disease association via the analysis of data sources, including patents, research publications, and clinical trial databases. The process revealed 20 novel targets for validation that Insilico narrowed down to the most promising candidate.
We then applied Chemistry42 to the chosen novel intracellular target. The platform uses generative and scoring engines to come up with hit compounds from scratch. All molecules created by Chemistry42 automatically have drug-like molecular structures and suitable physicochemical properties. The application of Chemistry42 to the novel target revealed by PandaOmics led to the generation of a library of small molecules.
Multiple molecules showed promising on-target inhibition, with one hit achieving nanomolar IC50 values without showing any sign of CYP inhibition. Optimization of that hit, named ISM001, improved solubility and resulted in good ADME properties. Subsequent studies found that these molecules improved fibrosis in a bleomycin-induced mouse lung fibrosis model and were safe when given to mice in a 14-day dose range-finding experiment.
After the positive preclinical studies, we initiated the first-in-human study in healthy volunteers to establish dose and basic safety which was concluded successfully. Phase 1 clinical trials are now underway in New Zealand and China.
Pharma Is Tapping Into AI Solutions
Since Chemistry42 launched, a number of pharmaceutical companies including nine of the top 30, have licensed the software and put it to work on their own pipeline programs. This includes Fosun Pharma, who nominated a preclinical candidate for the QPCTL program for cancer immunotherapy less than 40 days after partnering with Insilico, and EQRx, who partnered with Insilico last March.
Insilico Medicine’s drug discovery arm is also tapping into Pharma.ai’s capabilities with 30 internal pipeline programs in indications including cancer, inflammation, and COVID-19, moving toward clinical trials at a rapid pace. This is the real test of AI’s deep fake capabilities in the context of drug discovery. Not the quantities of predicted molecules it can produce, but the ability of those lead molecules to meet the safety and efficacy criteria of successful drugs and to withstand studies in animals and human patients. With Insilico’s end-to-end AI, the true potential of generative adversarial networks for transforming pharma is on the cusp of being realized.
Follow the Topic
-
Nature Biotechnology
A monthly journal covering the science and business of biotechnology, with new concepts in technology/methodology of relevance to the biological, biomedical, agricultural and environmental sciences.





Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in