AI Meets Antiviral: Engineering A Next-Generation Pan-Coronavirus Defense
Published in Chemistry, Microbiology, and Computational Sciences

The SARS-CoV-2 pandemic underscored the fragility of our therapeutic arsenal against emerging viral threats, especially those prone to rapid mutation. While spike protein-targeted mRNA vaccines and monoclonal antibodies provided critical short-term protection, their efficacy quickly waned as new variants emerged. The biological cat-and-mouse game of constantly updating strain-specific therapies underscored the need for therapeutic strategies that weren't tethered to the virus’s most mutable feature. Researchers from Guangzhou Medical University, Insilico Medicine, Guangzhou National Laboratory, and their collaborators have published their work in Nature Communications developing ISM3312, a novel small-molecule inhibitor of the coronavirus family main protease (Mpro) for the broad treatment of coronavirus infections.
Challenges in the Current Antiviral Landscape
The emergence and spread of COVID-19 prompted an unprecedented therapeutic discovery effort that yielded a number of effective treatments and prophylactic vaccines. However, the appearance of subsequent variants like Omicron demonstrated how even small changes in the targeted viral factor, the spike protein in the case of SARS-CoV-2, can render existing therapies less effective, highlighting the need for fundamentally different therapeutic strategies, for this disease and for potential future ones.
Additionally, the biomolecular nature of mRNA vaccines and antibody therapies render them less stable and requiring refrigeration and special care for storage, transport, and administration. They are therefore more difficult to effectively distribute globally, particularly in areas with limited or interrupted logistical capacity.
Small molecule drugs offer a compelling alternative: they are less affected by surface mutations, they have simpler oral administration protocols, and they are more stable in ambient conditions to ease storage and distribution. Yet, even our current small-molecule tools face serious limitations ranging from drug-drug interaction risks to pharmacokinetic inefficiencies and the ever-looming threat of acquired resistance.
The Case for Targeting Mpro
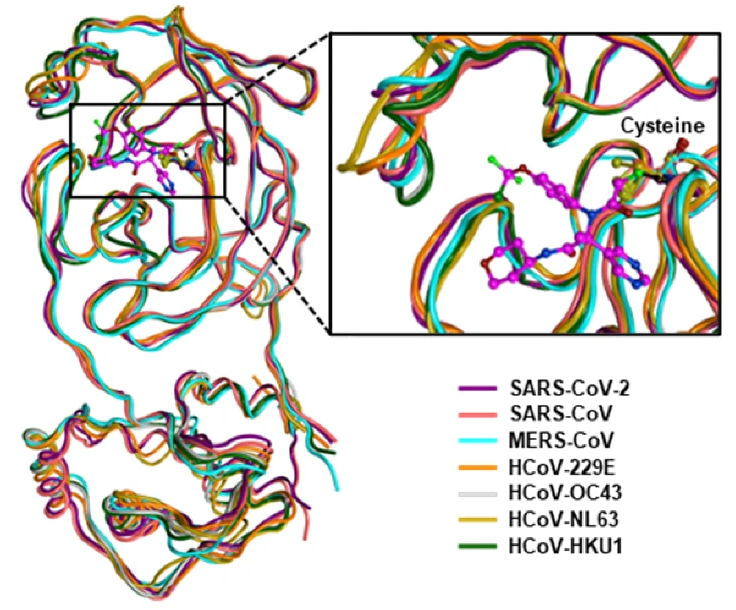
The SARS-CoV-2 main protease (Mpro) emerged early on as a prime drug target. Upon entry into host cells, two polyproteins translated from the SARS-CoV-2 genomic RNA encode 16 nonstructural proteins (nsp1-16) with various functions required for viral replication and transcription. Mpro is one of these nsps, itself undergoing autocleavage to release from the polyprotein and reach maturity, which then goes on to catalyze the release of other viral proteins from the polyprotein. Without Mpro, the virus cannot replicate. Importantly, this protease remains remarkably similar across human coronaviruses, from seasonal, lowly pathogenic 229E and OC43 to highly virulent MERS and SARS-CoV-2. The highly conserved Mpro active site across the range of human coronaviruses, down to individual catalytic amino acids like C145 and H41, makes it a remarkably attractive target for broad-spectrum antivirals.
Two Mpro inhibitors, Nirmatrelvir (one of two components of Paxlovid) and Ensitrelvir, have been approved for clinical use. But both carry liabilities. Both of these drugs are susceptible to efflux by P-glycoprotein (P-gp), a cellular pump that ejects drugs from the interior of cells, reducing intracellular concentrations and antiviral potency. To achieve therapeutic levels, Nirmatrelvir must be co-administered with ritonavir, an inhibitor of P-gp and CYP3A4 to boost oral exposure, which heightens the potential risk of drug-drug interactions and thus creates a litany of contraindicated drug interactions. Ensitrelvir, while not reliant on concentration-boosters, requires a high plasma concentration to achieve efficacy, which raises red flags about dose-limiting toxicity and practicality. Both drugs are also associated with acquired Mpro mutations that confer resistance.
A Rational Starting Point: Structure-Informed Drug Design
Our study set out to overcome these shortcomings. The goals were ambitious: identify a small molecule Mpro inhibitor that would (1) retain activity across coronavirus species, (2) function independently of PK boosters like ritonavir, (3) be less prone to resistance, and (4) maintain drug-like properties suitable for oral delivery and scalable manufacturing.
Choosing the right starting point is a critically important first step in structure-based drug design, as it informs every following iteration and molecular tweak, essentially restricting the chemical space available to probe. Rather than beginning with peptidomimetic inhibitors like Nirmatrelvir, which are generally less synthetically tractable and less well-suited for oral administration, the team prioritized small molecule scaffolds derived from known non-covalent small-molecule inhibitors of SARS-CoV Mpro, such as ML188 and X77, which were some of the few published coronavirus co-crystal structures available at the time. Crucially, these molecules engaged key residues in the Mpro binding pocket, including a critical hydrogen bond with His163, a moiety conserved in the S1 subpocket of the 3C-like proteases from rhinovirus to SARS-CoV-2 and an interaction target of Nirmatrelvir, a feature later encoded in the design criteria.
AI-Driven Design with Chemistry42
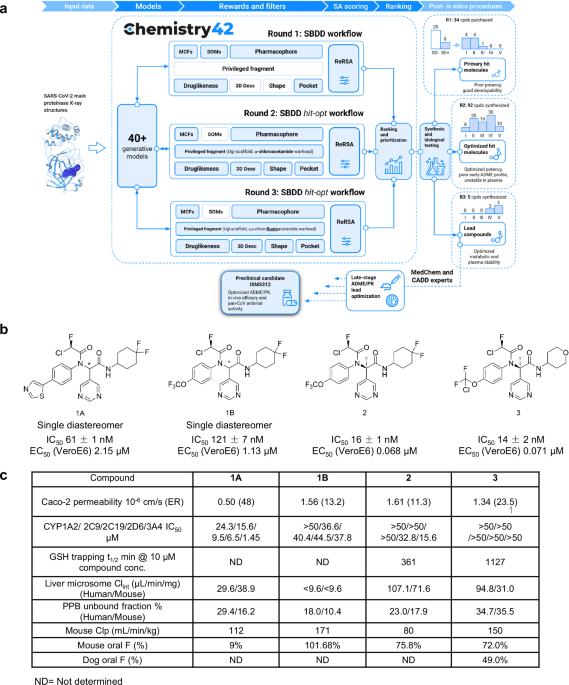
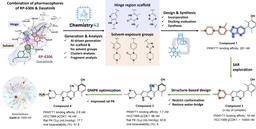
The design engine behind the discovery, Chemistry42, is an AI-powered platform that integrates generative chemistry with multi-objective optimization. The platform’s virtual screening pipeline was tasked with identifying compounds from established chemical libraries that maintained key pharmacophore interactions from the previously discovered inhibitors, kept important interactions within the binding pocket, optimized self-organizing maps derived from other protease inhibitor structures, and maximized synthetic accessibility. In vitro validation of the top predicted compounds identified a number of initial sub-micromolar hits featuring α-chloroacetamide warheads.
The AI-powered generative chemistry engine of Chemistry42 then took over to iteratively modulate the structure initiated by the top chemical library hit. The Pharmacophore module of Chemistry42 filled the many subpockets in the virtual Mpro structure with pharmacophore points responsible for favorable interactions, including an aromatic substructure to initiate pi-stacking to the His41 residue and two hydrophobic groups ensured complementarity to the hydrophobic S2 and S4 subpockets. A new warhead, α-chlorofluoroacetamide (CFA), then replaced the more reactive chloroacetamide to reduce covalent reactivity and improve its plasma stability. The next round of Chemistry42-generated structures and fine-tuning of key functional groups then yielded a suite of highly potent potential hits that were evaluated for efficacy in cellular models of infection and for pharmacokinetic properties (Figure 1).
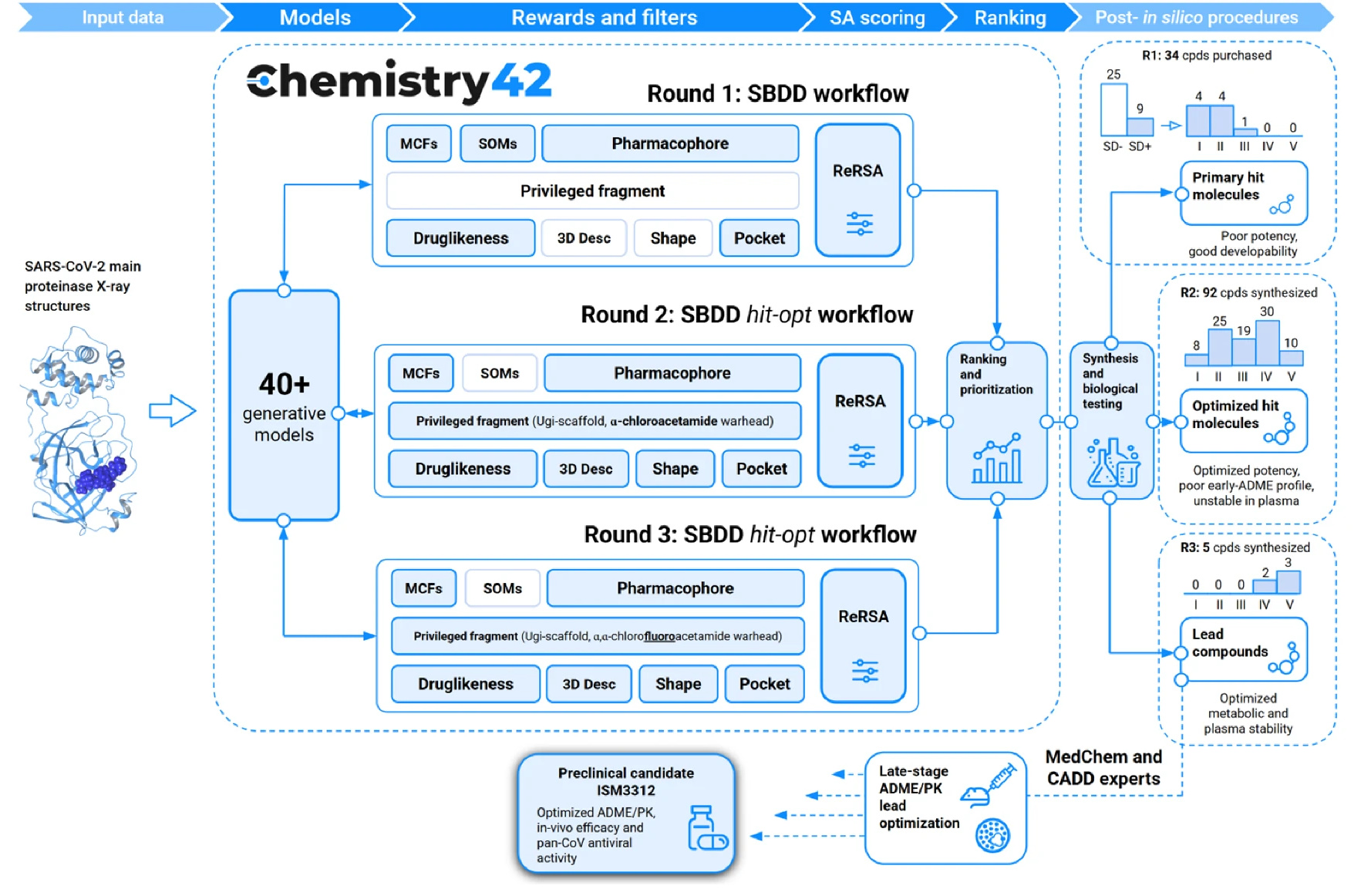
Figure 1. AI-powered Chemistry42 workflow for Mpro inhibitor discovery.
Meet ISM3312: A Covalent Game-Changer
The result was ISM3312, a covalent, irreversible Mpro inhibitor with a unique combination of properties.
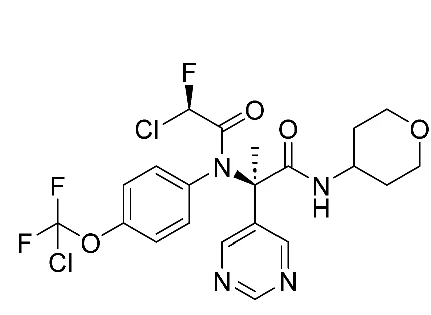
In enzymatic assays, ISM3312 exhibited very strong potency, with an IC50 of 14 nM against SARS-CoV-2 Mpro, as well as 33 nM for Omicron variant and 14 nM for SARS-CoV Mpro, similar to that of Nirmatrelvir and Ensitrelvir. But ISM3312 really stood out for its inhibitory activity against five other coronavirus Mpros, such as MERS and 229E, exhibiting an IC50 at least 3 to 25 times lower than the other Mpro inhibitors across the board (Ensitrelvir not showing any inhibitory activity against several of them) (Figure 2). Inhibition by ISM3312 also did not diminish over time, in contrast to the other Mpro inhibitors, which suggests its irreversible, covalent inhibition yields strong inhibition potential over longer periods than the other, reversible inhibitors.
ISM3312 was also rigorously tested in vitro in cell cultures infected with coronavirus, in organoids modeling the human upper respiratory tract, and in mouse models engineered to support coronavirus infection (i.e., expressing ACE2 or APN receptors). It again showed potent antiviral activity against a spectrum of CoVs, including all tested SARS-CoV-2 variants, MERS-CoV, and seasonal CoVs like OC43 and 229E.
It should not be overlooked that optimizing molecular design for synthetic accessibility allowed ISM3122 to be synthesized from readily available and inexpensive starting materials in a simple, two-step reaction. In addition, high yield and purity (>98%) achieved without chromatographic purification enabled easy scaling up from benchtop to kilogram-scale synthesis, making it an ideal candidate for rapid deployment in emergency scenarios.
with Mpro from other human coronaviruses
Meeting the Challenges of Mpro inhibition
Nirmatrelvir’s efficacy in particular is compromised by cellular export by the P-gp efflux pump, which necessitates coadministration with the P-gp–inhibiting ritonavir to achieve therapeutic intracellular concentrations. ISM3312’s efficacy, however, is not compromised by cellular efflux mechanisms, as it had a lower EC50 as a monotherapy compared to Nirmatrelvir and Ensitrelvir and with minimal benefit conferred by coadministration with a P-gp inhibitor across SARS-CoV-2 variants. Administration as a monotherapy would greatly simplify the formulation processes and logistics involved in potential global emergencies requiring rapid manufacturing and distribution.
Resistance evolution is also a major concern for any targeted antiviral therapy. In 18-passage resistance assays, induced only modest resistance (15-fold EC50 shift), compared to Nirmatrelvir's 153-fold increased EC50. Intriguingly, the resistance mutations that appeared more in ISM3312-resistant viruses than in Nirmatrelvir- or Ensitrelvir-resistant viruses were largely distal to the protease inhibitor binding site. Biochemical assays showed these did not impact enzymatic inhibition directly, suggesting an indirect resistance mechanism, possibly by altering Mpro’s interaction with its substrate. This opens new research directions to decode noncanonical mechanisms of resistance and to explore rational combinations (e.g., ISM3312 + Nirmatrelvir) that might slow resistance even further.
Importantly, ISM3312 retained potency against most Nirmatrelvir- and Ensitrelvir-resistant Mpro mutants. Structural modeling suggested a weaker but broader binding mode that makes ISM3312 less vulnerable to destabilizing mutations, especially those that severely disrupt hydrogen bonding networks critical for Nirmatrelvir. This may be where the irreversibly covalent activity of ISM3312 confers great advantage: a slightly lower binding affinity that actually helps overcome resistance mutations is compensated with longer-lasting irreversible Mpro inhibition.
A Step Toward Pandemic Preparedness
ISM3312 is now undergoing further development as a broad-spectrum antiviral candidate, entering phase 1 clinical trials (CTR20230768). Its ability to overcome limitations of existing drugs, coupled with a high barrier to resistance, makes it a promising agent not just for current CoVs, but as a frontline defense against future outbreaks. Combined with the ease of manufacture (successfully synthesized at kilogram-scale from inexpensive starting components), these features perhaps position it as one tool well-suited to be “prepped and ready” in a strategic reserve.
From conception to optimization, this study exemplifies the convergence of AI-driven discovery and classical pharmacology. As we face a future of unpredictable viral emergence, we believe this approach can serve as a model for rapid, rational drug development, moving us one step closer to a more resilient global health infrastructure.
Follow the Topic
-
Nature Communications
An open access, multidisciplinary journal dedicated to publishing high-quality research in all areas of the biological, health, physical, chemical and Earth sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Women's Health
Publishing Model: Hybrid
Deadline: Ongoing
Tumor Microenvironment Crosstalk and Therapeutic Implications
Publishing Model: Hybrid
Deadline: Nov 02, 2026



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in
Related:
◾ AI in antiviral drug discovery