Altered chromatin topologies caused by balanced chromosomal translocation lead to circular iris coloboma
Published in Genetics & Genomics
Background
With the increasing clinical implementation of next-generation sequencing, the gene diagnostic yield for monogenic diseases has improved but still rarely exceeds 50%1-3. For the majority of unsolved patients, this may be explained by limited power to resolve complex structural variations (SVs) of the short-read sequencing platform and incomplete knowledge of the functional consequences of most noncoding variants4. Although long-read sequencing technology improves the identification of complex SVs, the analytical challenge around variant interpretation persists, especially for balanced SVs with noncoding breakpoints5-7. Noncoding regions, representing approximately 98% of the human genome, have recently attracted attention in relation to genetically unsolved patients with Mendelian disorders8-10. To date, a number of noncoding variants have been identified, most of which were recognized according to a physical location in or adjacent to the known disease-causing gene, such as intronic, regulatory, upstream or downstream regions of known genes. These noncoding variants are reasonably linked with the aberrant expression of their adjacent known genes11,12. However, for variants at intergenic regions, evidence, both at the genetic level and in functional analysis, is necessary to confirm the pathogenicity of these variants.
Key findings
Identification of a unique condition of central iris hypoplasia in a large family
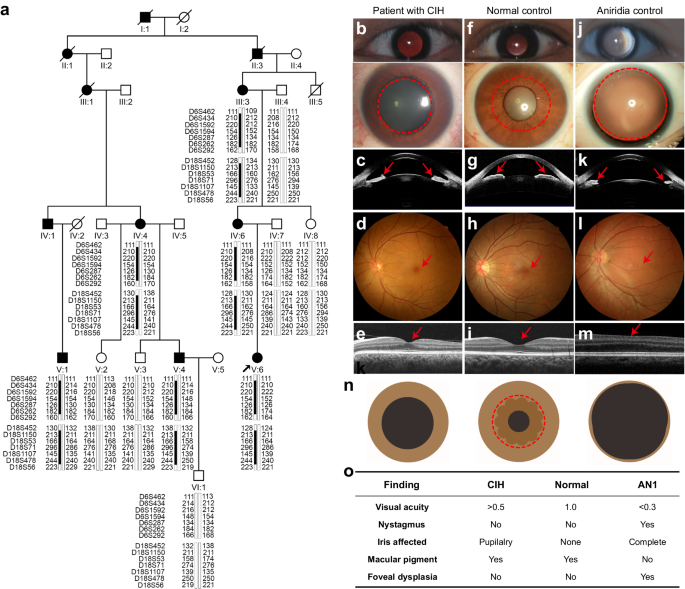
In our study, we identified the central iris hypoplasia (CIH) as a unique condition in a large family (Figure 1a). All the six affected patients of the large family had a history of photophobia since birth and a central absence of the iris pupillary zone alone in both eyes with a pseudo-enlarged pupil. They had clear cornea and normal-like fundus with foveal reflex without nystagmus or any other obvious abnormality. The CIH in this family is different from aniridia, which is characterized by iris coloboma involving both pupillary and peripheral zones, foveal hypoplasia, congenital nystagmus, and low visual acuity (Figure 1b).
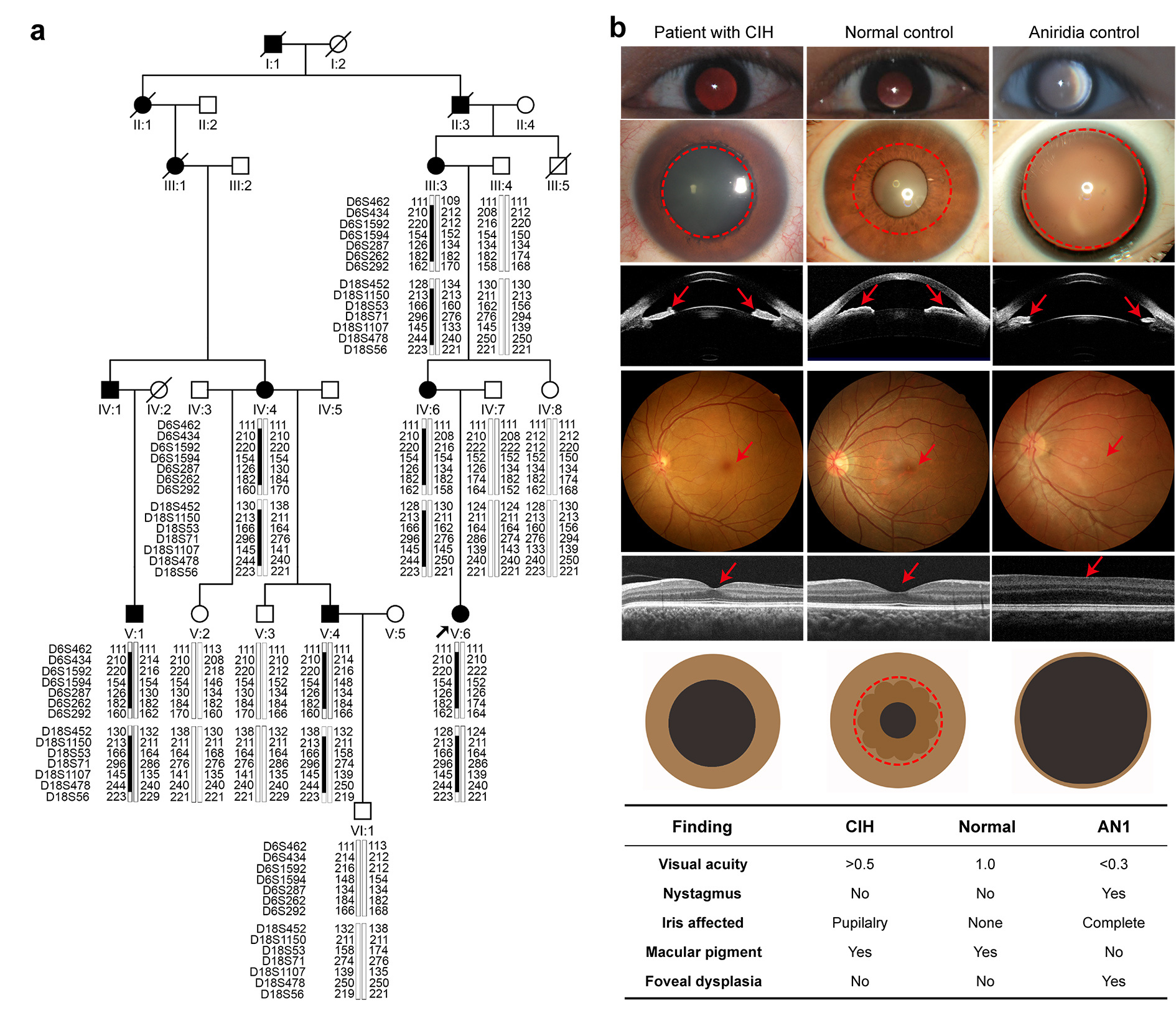 Figure 1. The CIH phenotype in a large family
Figure 1. The CIH phenotype in a large family
Identification of a balanced translocation with intergenic breakpoints in the family
A genome-wide linkage scan was performed on genomic DNA from all 12 individuals, and two-point linkage analysis mapped two loci on chromosomes 6q15-q23.3 and 18.p11.31-q12.1 (Figure 1a). After short-read sequencing excluded SNVs and InDels inside the linkage intervals, long-read genome sequencing identified a balanced translocation t (6;18) (q22.31;p11.22) with both breakpoints at intergenic regions, chr6:121619304 and chr18:10432739 (Figure 2a). Sanger sequencing confirmed that the noncoding translocation variation completely co-segregated with CIH within the family, further supporting it as the disease-causing change for this family (Figure 2b).
The translocation led to reorganizations of chromatin domains over the breakpoints
To further explore the molecular mechanism underlying CIH caused by the balanced structural variation, we generated induced pluripotent stem cells (iPSCs) from peripheral blood mononuclear cells of one patient and one unaffected individual in the family. Hi-C sequencing was conducted on the iPSCs samples from the two individuals, and it showed that the translocation disrupted the two chromatin topologically associating domains (TADs) on chromosome 6 and 18 in the normal control but led to the formation of neo-TADs over the breakpoints on the derivative chromosomes in the patient (Figure 2c). CUT&Tag analysis identified the presence of a tissue-specific enhancer cluster on chromosome 6, which was driven next to the APCDD1 promoter in a neo-TAD by the translocation (Figure 2c). RNA-seq analysis identified that APCCD1 was the only gene with a statistically significant expression change among 40 genes within 2.5 Mb of each breakpoint (Figure 2d). These findings suggest that the translocation rewired the three-dimensional chromatin structure, leading to aberrant interact between enhancers and APCDD1 promoter and thereby upregulating its expression.
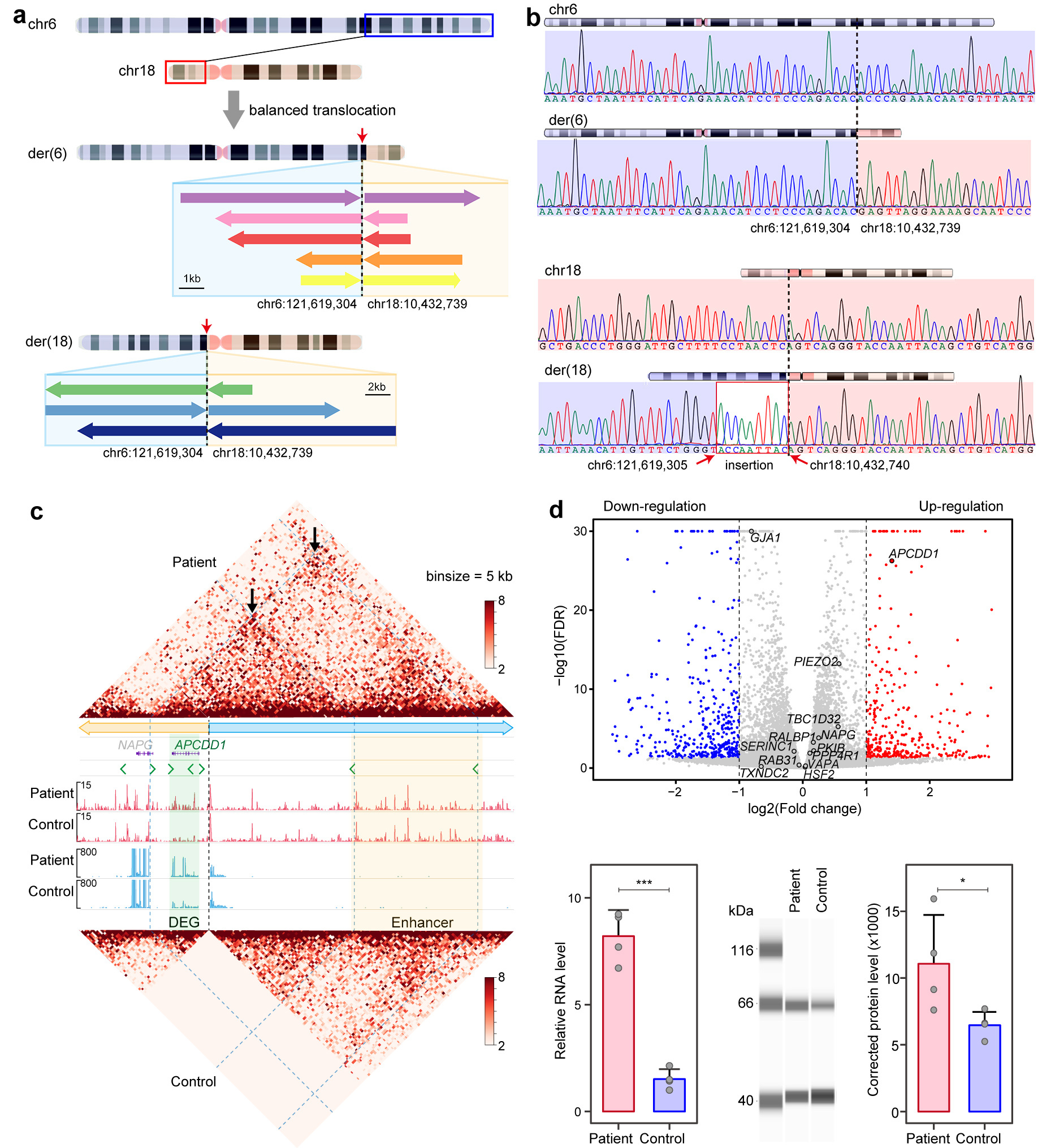 Figure 2. Noncoding translocation generated neo-TAD and upregulated APCDD1 expression
Figure 2. Noncoding translocation generated neo-TAD and upregulated APCDD1 expression
APCDD1 play a role in development and maintain of the iris
To assess the potential roles of APCDD1 in the iris, we conducted immunofluorescence staining in eyes of human adult as well as mice at different stages. The results demonstrated that APCDD1 was specifically expressed in the fibroblasts of the human iris (Figure 3a), whereas the expression pattern in mouse eyes transferred from the peripheral inner layer of the neural retina (E11.5 ~ E14.5) to constriction in the boundary of the ciliary body and the retina (E17.5 ~ P3M) (Figure 3b). These results confirmed that APCDD1 plays a role in development and maintenance of the iris.
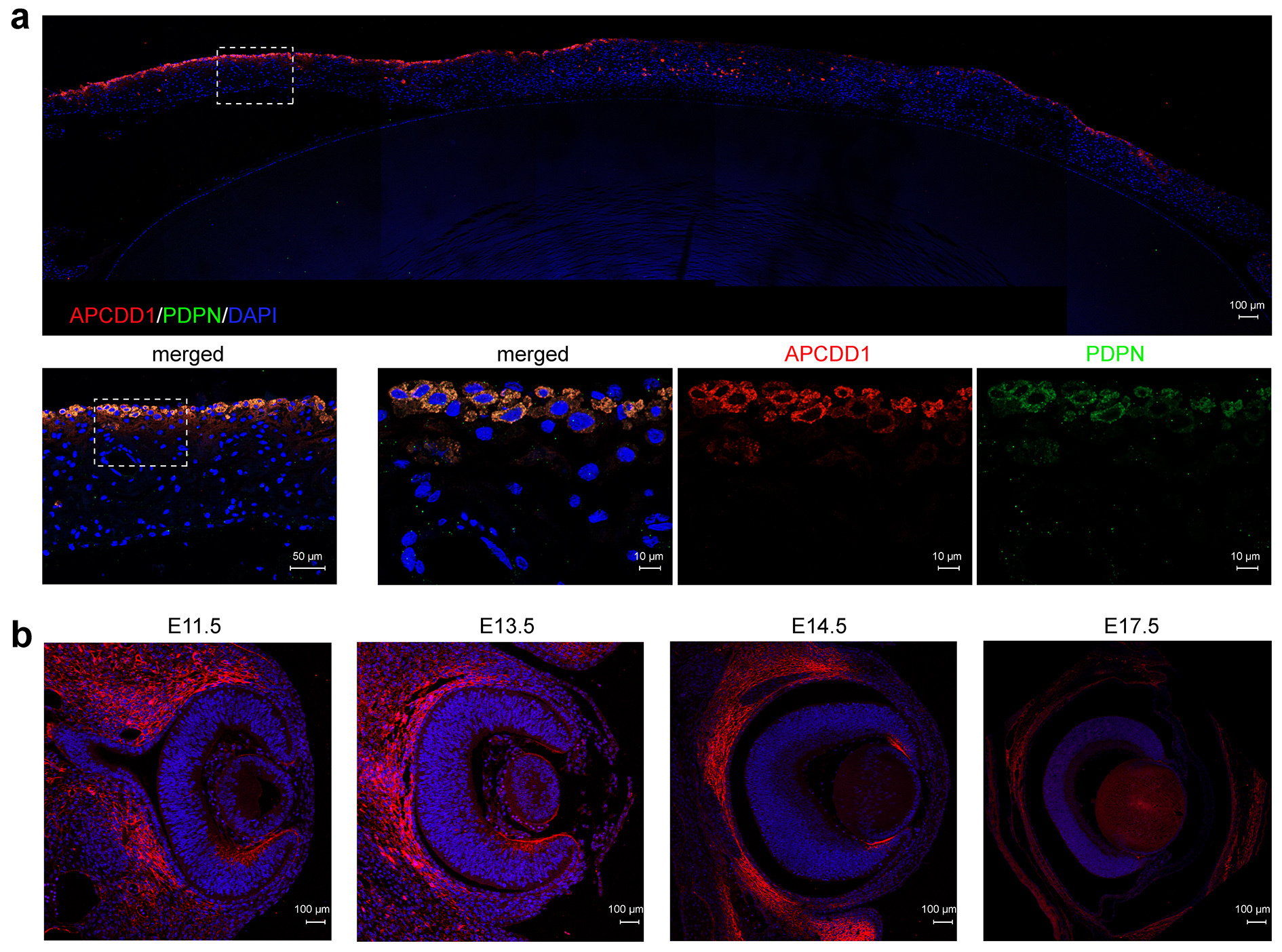 Figure 3. Expression pattern of APCDD1 in the human iris and embryonic mouse eyes
Figure 3. Expression pattern of APCDD1 in the human iris and embryonic mouse eyes
Conclusion
Our research underscores several implications in the area of medical genetics. First, we provide evidence for the identification of complex SVs at noncoding intergenic regions relevant to Mendelian diseases. Although such complex SVs can be identified by cytogenetic methods, they are rarely concerned in routine genetic diagnosis for Mendelian diseases13. A uniform phenotype in large families with definite mapped loci based on genome-wide linkage analysis provides unique opportunities to explore noncoding SVs for Mendelian diseases, as in the current study’s analysis of the large family with CIH. Second, pathogenicity of noncoding SVs is achieved through disrupting the three-dimensional chromatin structure14,15, which provide an avenue to interpreting the function and pathogenicity of noncoding variants, such as the balanced translocation with noncoding breakpoints in this study. Third, the discovery of disease-associated genes via increased dosage effects has been particularly challenging, as in the case of APCDD1 in this study. The majority of genes causing human diseases are identified as a result of loss-of-function variants and occasionally via gain-of-function16,17. However, variants with increased dosage effects are easily missed by current variant prioritization strategies in clinical genetic pipelines18. Additional studies aimed at understanding the upregulation of functional genes through coding or noncoding elements, as in the current study, may improve the prediction probabilities for variants with an effect on the upregulation of the genes that contribute to human diseases, which may in turn elucidate the genetic cause of diseases in approximately half of unsolved patients with inherited disease17,19,20.
References
- Investigators, G.P.P. et al. 100,000 Genomes Pilot on Rare-Disease Diagnosis in Health Care - Preliminary Report. N Engl J Med 385, 1868-1880 (2021).
- Turro, E. et al. Whole-genome sequencing of patients with rare diseases in a national health system. Nature 583, 96-102 (2020).
- Alix, T. et al. Predictors of the utility of clinical exome sequencing as a first-tier genetic test in patients with Mendelian phenotypes: results from a referral center study on 603 consecutive cases. Hum Genomics 17, 5 (2023).
- Taylor, J.C. et al. Factors influencing success of clinical genome sequencing across a broad spectrum of disorders. Nat Genet 47, 717-726 (2015).
- Collins, R.L. et al. A structural variation reference for medical and population genetics. Nature 581, 444-451 (2020).
- Audano, P.A. et al. Characterizing the Major Structural Variant Alleles of the Human Genome. Cell 176, 663-675 e19 (2019).
- Sanchis-Juan, A. et al. Complex structural variants in Mendelian disorders: identification and breakpoint resolution using short- and long-read genome sequencing. Genome Med 10, 95 (2018).
- Valente, E.M. & Bhatia, K.P. Solving Mendelian Mysteries: The Non-coding Genome May Hold the Key. Cell 172, 889-891 (2018).
- Filatova, A. et al. Annotation of uORFs in the OMIM genes allows to reveal pathogenic variants in 5'UTRs. Nucleic Acids Res 51, 1229-1244 (2023).
- Wakeling, M.N. et al. Non-coding variants disrupting a tissue-specific regulatory element in HK1 cause congenital hyperinsulinism. Nat Genet 54, 1615-1620 (2022).
- Fresard, L. et al. Identification of rare-disease genes using blood transcriptome sequencing and large control cohorts. Nat Med 25, 911-919 (2019).
- Yepez, V.A. et al. Clinical implementation of RNA sequencing for Mendelian disease diagnostics. Genome Med 14, 38 (2022).
- Tommerup, N. Mendelian cytogenetics. Chromosome rearrangements associated with mendelian disorders. J Med Genet 30, 713-27 (1993).
- Lupianez, D.G. et al. Disruptions of topological chromatin domains cause pathogenic rewiring of gene-enhancer interactions. Cell 161, 1012-1025 (2015).
- de Bruijn, S.E. et al. Structural Variants Create New Topological-Associated Domains and Ectopic Retinal Enhancer-Gene Contact in Dominant Retinitis Pigmentosa. Am J Hum Genet 107, 802-814 (2020).
- Matharu, N. & Ahituv, N. Modulating gene regulation to treat genetic disorders. Nat Rev Drug Discov 19, 757-775 (2020).
- Zschocke, J., Byers, P.H. & Wilkie, A.O.M. Mendelian inheritance revisited: dominance and recessiveness in medical genetics. Nat Rev Genet 7, 442-463 (2023).
- Gerasimavicius, L., Livesey, B.J. & Marsh, J.A. Loss-of-function, gain-of-function and dominant-negative mutations have profoundly different effects on protein structure. Nat Commun 13, 3895 (2022).
- Sopko, R. et al. Mapping pathways and phenotypes by systematic gene overexpression. Mol Cell 21, 319-30 (2006).
- Collins, R.L. et al. A cross-disorder dosage sensitivity map of the human genome. Cell 185, 3041-3055 e25 (2022).
Follow the Topic
-
Nature Communications
An open access, multidisciplinary journal dedicated to publishing high-quality research in all areas of the biological, health, physical, chemical and Earth sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Women's Health
Publishing Model: Hybrid
Deadline: Ongoing
Tumor Microenvironment Crosstalk and Therapeutic Implications
Publishing Model: Hybrid
Deadline: Nov 02, 2026




Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in